What Can DP Do too? | Using Machine Learning to Explore the Catalytic Kinetics of Metal Nanoclusters under Confinement Conditions
On October 17, 2024, the research paper titled "Entropy in catalyst dynamics under confinement" by the AI4EC Lab/Professor Jun Cheng's research group from Xiamen University was published online in the international journal Chem. Sci. The first author of the paper is Qiyuan Fan(currently a teacher at the School of Chemistry and Chemical Engineering, Shanxi University). This work was completed under the guidance of Professor Jun Cheng and with the guidance and support of Academician Zhongqun Tian, Academician Xinhe Bao from the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Academician Weinan E from Peking University, and Professor Ye Wang from Xiamen University.
Research Background
In heterogeneous catalytic reactions, the active sites of the catalyst are crucial in determining the catalytic performance. Traditionally, active sites are often regarded as stable structures with specific atomic arrangements. However, in recent years, an increasing number of studies have found that the dynamic behavior of catalysts under reaction conditions also significantly affects catalytic activity. Particularly in spatially confined conditions, such as when metal nanoclusters are encapsulated in the pores of carbon nanotubes, zeolites, or metal-organic frameworks, the confinement effect has a profound impact on the structure and kinetic properties of the catalyst, thereby altering the catalytic performance.
Nevertheless, previous studies have mostly focused on static geometric optimization, ignoring the dynamic behavior of catalysts and the corresponding entropy effects in confined environments. The structure of the catalyst continuously changes during the reaction process and may form a series of metastable states, which play an important role in catalyzing chemical reactions. Therefore, understanding the structural dynamics and entropy changes of catalysts under confinement effects is essential for revealing the mechanism of catalytic reactions.
This study, by combining DeePMD-kit, for the first time explores the entropy effect of metal nanoclusters under confinement conditions and its impact on catalytic reactions from the perspectives of kinetics and thermodynamics. This work provides a new perspective for in-depth understanding of the dynamic confinement effect and contributes to promoting research on the design and optimization of nanocatalysts in complex chemical systems.
Methods
The methods section of this article mainly includes the following contents:
Training of Machine Learning Potential (MLP): Using the Deep Potential Generator (DP-GEN) package, a series of iterative steps including exploration, annotation, and training were employed. During the training process, the network structure parameters of MLP were (25, 50, 100) and (240, 240, 240), and the learning rate started from 10−3 and gradually exponentially decayed to 10−8. The cutoff radius was set to 7 Å. Each model underwent multiple training steps, and the final model had over 4,000,000 training steps.
Machine Learning Molecular Dynamics (MLMD): The exploration step was based on the LAMMPS code, and the initial training data was composed of the initial and final state structures randomly selected from the 5 ps AIMD trajectory. To calculate the reaction free energy, a series of constrained molecular dynamics simulations were performed along the preset reaction coordinate (such as the O-O bond length) to sample structures at different temperatures.
Annotation Step: In the exploration step, the model deviation of σmax(f) was used as the error indicator to select the structures that needed annotation. The annotation process used DFT calculations (completed by VASP) and added these calculation results to the data set.
DFT Calculation: VASP was used for energy and force calculations during the annotation process, employing the PAW method and PBE functional. Different energy and force convergence criteria were set in the calculation, and the simulated system was placed in a specific-sized cubic box.
Free Energy Calculation: The final free energy calculation was performed through the modified CP2K interfaced with DeepMD-kit. Based on the trained MLP model, the free energy curve was obtained by calculating the average force using the Lagrange multiplier algorithm. The simulation temperature range was from 200 K to 1200 K, and the free energy under the O-O bond length was calculated through constrained MD.
Estimation of Statistical Error: By dividing the equilibrated MLMD trajectory into five time periods, the force and free energy were independently calculated for each period to evaluate the error of the calculation results at each O-O bond length.
Results and Discussion
MLP Accelerates Free Energy Calculation: Efficient and Accurate Simulation of O₂ Dissociation Reaction
Figure 1 shows the overall workflow of the MLP-accelerated free energy calculation method and the comparison between the generated MLP model and the results of first-principles calculations (DFT). In Figure 1A, the working steps of the MLP model are detailed, including how to use the machine learning potential model to efficiently perform molecular dynamics simulations, thereby significantly reducing the computational cost. Figures 1B and 1C compare the atomic forces and energies under the O-O bond length calculated by the MLP model and DFT at different temperatures, respectively. The results show good agreement between the two at different temperatures. The insets give the errors of atomic forces (in eV/Å) and energies (in eV), and these validation data indicate that the MLP model has reliable accuracy and can be used for further kinetic and thermodynamic analyses.
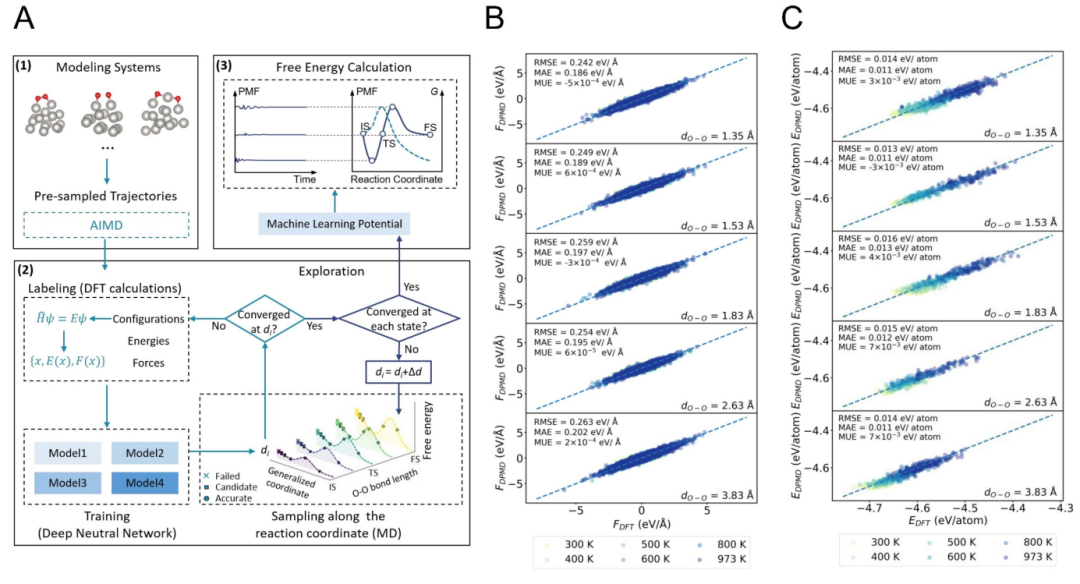
Figure 1 (A) Schematic diagram of the workflow of the MLP-accelerated free energy calculation method. (B) Comparison of atomic forces under the O-O bond length calculated by the generated MLP model and DFT at different temperatures. (C) Comparison of energies under the O-O bond length calculated by the MLP model and DFT at different temperatures. The insets show the errors of atomic forces (in eV/Å) and energies (in eV) used for validating the data.&
O₂ Dissociation in Pt Nanoclusters: Influence of Confinement Effect on Free Energy and Entropy Change
Figure 2 shows the dissociation free energy and structural schematic of O₂ on Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT calculated by MLMD (machine learning accelerated molecular dynamics). Figures 2A and 2B are typical structural snapshots of O₂ adsorbed on Pt₁₅@CNT and Pt₁₅/CNT, respectively, where silver and red spheres represent Pt atoms and O atoms, respectively. Figure 2C shows the relationship between the dissociation reaction free energy (∆rG) of O₂ in these three systems and temperature, and the fitted curve indicates the trend of free energy change in different systems. Figure 2D shows the change of reaction entropy (∆rS) during the O₂ dissociation process with temperature, reflecting how the confined environment affects the kinetics and thermodynamics of the reaction.
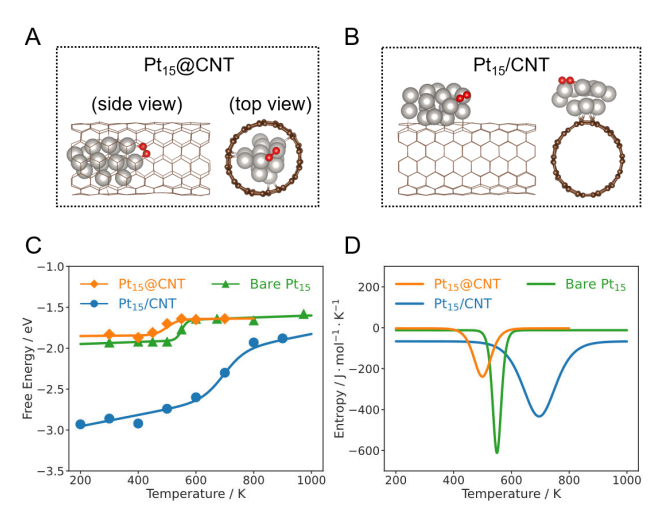
Figure 2 Dissociation free energy of O₂ on Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT calculated based on MLMD. Typical structural snapshots of O₂ adsorbed on Pt₁₅@CNT (A) and Pt₁₅/CNT (B). Silver and red spheres represent Pt atoms and O atoms, respectively. (C) Relationship between the dissociation reaction free energy (∆rG) of O₂ on Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT and temperature. The solid line is the fitted curve. (D) Relationship between the dissociation reaction entropy change (∆rS) of O₂ on Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT and temperature.
Analysis of the Influence of Confined Environment on the Free Energy and Entropy Effect of O₂ Dissociation in Pt₂₇ Nanoclusters
Figure 3 shows the structural and thermodynamic characteristics of the O₂ dissociation reaction on Pt₂₇@CNT and bare Pt₂₇. Figure 3A is a typical structural snapshot of O₂ adsorbed on Pt₂₇@CNT, where silver and red spheres represent Pt atoms and O atoms, respectively. Figure 3B shows the relationship between the dissociation reaction free energy (∆rG) of O₂ on Pt₂₇@CNT and bare Pt₂₇ and temperature, and the fitted curve shows the trend comparison between the two systems. Figure 3C shows the change of reaction entropy (∆rS) during the O₂ dissociation process on Pt₂₇@CNT and bare Pt₂₇ with temperature, from which the influence of the confined environment on the entropy effect of the reaction can be seen. Through these analyses, the study reveals the differences in reaction free energy and entropy change between Pt₂₇@CNT and bare Pt₂₇, providing a new perspective for understanding the catalytic performance under confinement conditions.
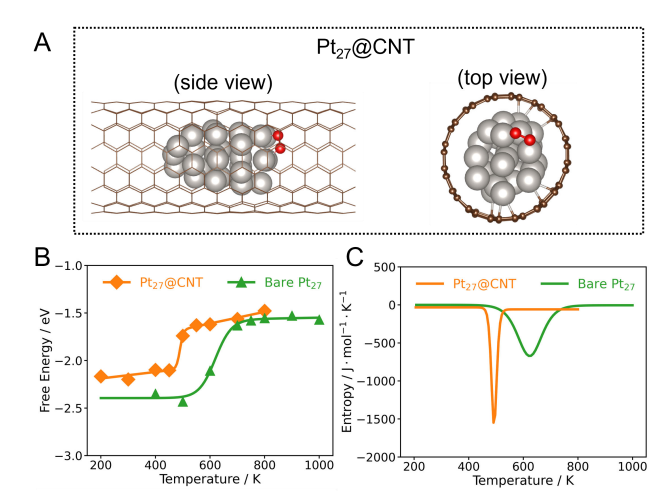
Figure 3 (A) Typical structural snapshot of O₂ adsorbed on Pt₂₇@CNT. Silver and red spheres represent Pt atoms and O atoms, respectively. (B) Relationship between the dissociation reaction free energy (∆rG) of O₂ on Pt₂₇@CNT and bare Pt₂₇ and temperature. The solid line is the fitted curve. (C) Relationship between the dissociation reaction entropy change (∆rS) of O₂ on Pt₂₇@CNT and bare Pt₂₇ and temperature.
Phase Transition Behavior and Structural Stability of Pt₁₅ Clusters under Confinement Conditions: Interpretation from RMSD Analysis
Figure 4 shows the phase transition behavior and structural stability of Pt₁₅@CNT, Pt₁₅/CNT, and bare Pt₁₅ in different states. Figures 4A and 4B show the phase transition behaviors of these three systems in the initial state (IS) and final state (FS), respectively. The calculated values are represented by points, and the fitted curves are represented by lines, revealing how the confinement conditions affect the phase transition process. Figure 4C shows the probability density of the RMSD of the Pt atom positions in the final state (FS) of Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT. The RMSD is calculated from the molecular dynamics (MD) trajectory relative to the initial frame and is used to characterize the volatility of the atom positions and the structural stability of the system. Through these analyses, the study reveals how the confined environment affects the structural stability and phase transition behavior of platinum clusters, providing important information for understanding the microscopic mechanism of confined catalysis.
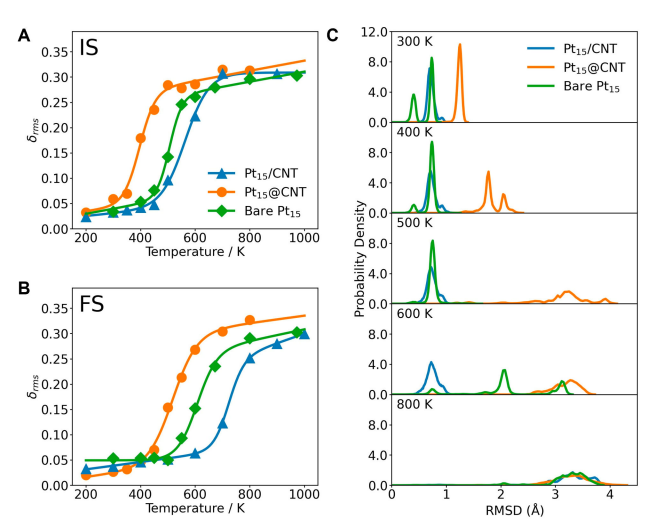
Figure 4 Phase transition behaviors of Pt₁₅@CNT, Pt₁₅/CNT, and bare Pt₁₅ in the initial state (IS, A) and final state (FS, B). The calculated values are represented by points, and the corresponding fitted curves are represented by lines. (C) Probability density of the RMSD of the Pt atom positions of Pt₁₅@CNT, bare Pt₁₅, and Pt₁₅/CNT in the final state (FS) at different temperatures. The RMSD of the Pt atom positions is calculated from the MD trajectory relative to the first frame.
Conclusions
In this study, the research team utilized machine learning-accelerated molecular dynamics (MLMD) and free energy calculations to explore the catalytic kinetics under confinement conditions from the perspectives of entropy effects and structural dynamics for the first time. The research shows that in the confined environment, the entropy effect of the catalyst significantly affects its activity, and the confinement effect can enhance the structural dynamics of the cluster, lower the melting point, and thus promote the reaction to occur at a lower temperature, preventing the catalyst from forming unfavorable oxides. Through detailed analyses of free energy, entropy, and structural dynamics, this study reveals how confinement conditions change the phase transition behavior and reaction mechanism of the catalyst, providing a new perspective for understanding the microscopic mechanism of confined catalysis and will inspire more in-depth research on dynamic catalysis under confinement in the future.