GPUMD&NEP Helps Decode the Secrets of Solid-State Electrolytes - How Does Lithium Nonstoichiometry Affect the Performance of Li7La3Zr2O12?
GPUMD is an efficient domestic molecular dynamics simulation software developed and maintained by Professor Zheyong Fan from Bohai University. The software first released its public version 1.0 in 2017 [Computer Physics Communications 218, 10 (2017)] and has currently been iterated to version 3.9.4. GPUMD includes both commonly used empirical potentials and NEP (Neuroevolution Potential) machine learning potentials. Up to now, GPUMD has been used by thousands of users in many countries around the world and has attracted dozens of researchers to participate in its development. It is widely applied in fields such as heat and mass transfer, mechanical properties, structural phase transitions, irradiation damage, spectroscopy, and catalysis. Related achievements have been published in top academic journals such as Nature, Nature Communications, J. Am. Chem. Soc, ACS Nano, Phys. Rev. Lett, J. Mech. Phys. Solids, J. Chem. Theory Comput., Phys. Rev. B, and J. Chem. Phys.
In June 2024, GPUMD&NEP joined the DeepModeling community. As an innovative and highly efficient MD simulation and machine learning potential function tool, it further provides support for the Materials Genome Project and the AI4S community.
Introduction
In recent years, all-solid-state lithium-ion batteries have attracted much attention due to their high safety and high energy density. As a solid-state electrolyte material with high ionic conductivity and stability, Li7La3Zr2O12 (LLZO) is particularly remarkable. However, there is a significant difference between the theoretically predicted activation energy (about 1.2 eV) and the experimentally measured value (about 0.45 eV) for lithium-ion migration in the tetragonal phase LLZO. This contradiction limits the in-depth understanding and optimization of the material's performance.
Recently, Professor Yizhou Zhu's team from Westlake University successfully solved this problem by using GPUMD&NEP and achieved high-precision and large-scale simulations of lithium-ion migration in LLZO. The research accurately predicted key properties such as the temperature of the tetragonal-cubic phase transition, lattice parameters, ionic conductivity, activation energy for lithium-ion migration, and defect concentrations of lithium and oxygen in LLZO from the theoretical level, and these results are almost completely consistent with the experimentally measured values. Notably, this is also the first application of GPUMD&NEP in the field of solid-state electrolytes, demonstrating its strong potential in studying large-scale complex material systems.
GPUMD&NEP: A Perfect Combination of High Efficiency and High Precision
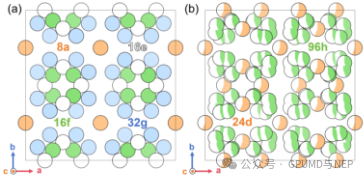
Figure 1. (a) Lithium sublattice in tetragonal phase and (b) cubic phase LLZO.
The unit cell of LLZO contains 192 atoms, and the LaO8 and ZrO6 polyhedra form a three-dimensional framework that supports the lithium-ion transport network. Its lithium sublattice shows significant differences between the tetragonal phase (t-LLZO) and the cubic phase (c-LLZO): in the tetragonal phase, lithium ions vibrate between ordered crystal sites with limited diffusion, while the disordered sublattice structure in the cubic phase significantly enhances the ion migration ability. There have been some previous molecular dynamics simulation studies on this system. However, classical molecular dynamics simulations based on the Buckingham potential lack sufficient precision, and expensive ab initio molecular dynamics simulations can only perform short-time and small-scale simulations (usually 1×1×1 unit cell). More importantly, previous studies have shown that there are significant differences between the simulation results of unit cells and supercells, and there are obvious deviations in the described activation energy for lithium-ion diffusion.
GPUMD&NEP perfectly solves this problem. As a machine learning force field that balances precision, efficiency, and cost, NEP accurately predicts this complex solid-state electrolyte model using only 2024 structures. The training set includes a total of 2024 structures collected through random perturbations, strains, and active learning strategies. It contains 1978 intrinsic LLZO structures and 46 structures with Li-O defects. The root mean square errors of NEP in predicting the energy, force, and stress of the training set are 0.66 meV/atom, 64.88 meV/Å, and 0.0767 GPa, respectively. In addition, with the support of GPUMD, researchers can break through the bottlenecks of size and simulation time, conduct high-precision long-time simulations at low cost, and accurately reproduce key experimental values such as the activation energy for lithium-ion migration, defect concentration, and ionic conductivity in LLZO, fully demonstrating the great potential of GPUMD&NEP in the field of solid-state electrolytes.
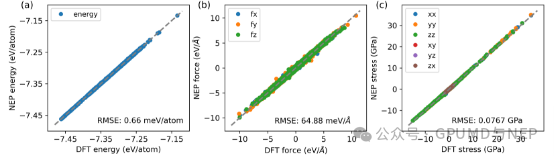
Figure 2. Comparison of (a) energy, (b) force, and (c) stress values predicted by NEP with DFT calculation results.
The following are the core findings and breakthroughs in the research:
Point 1: Accurately Describe the Tetragonal-Cubic Phase Transition
The tetragonal phase LLZO undergoes a phase transition at about 900 K to form a cubic phase with high conductivity (Figure 3). The calculated lattice parameters are highly consistent with the experimentally measured results (purple open circles). GPUMD&NEP accurately describes the phase transition behavior of LLZO, further verifying the reliability of this method.
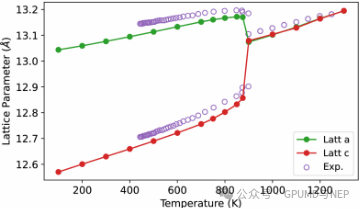
Figure 3. Evolution of the calculated LLZO lattice parameters. Purple open circles are the experimental lattice parameters.
Point 2: Significant Influence of Lithium Nonstoichiometry on Ion Migration
The research found that the introduction of a small amount of lithium defects significantly reduces the activation energy for ion migration. In particular, after introducing about 5.2‰ oxygen defects, a small number of lithium vacancies are generated in the system to maintain charge balance. This combination of lithium and oxygen defects reduces the activation energy from the theoretical value of 1.227 eV to 0.425 eV, which is almost completely consistent with the experimentally measured value of 0.41 - 0.45 eV (Figure 4). At the same time, the ionic conductivity is increased by about 10 orders of magnitude, which is also highly consistent with the experimental results. Surprisingly, the concentration of oxygen defects in the simulation is also highly consistent with the oxygen defect concentration of about 5‰ in single-crystal LLZO reported by Kubicek et al.
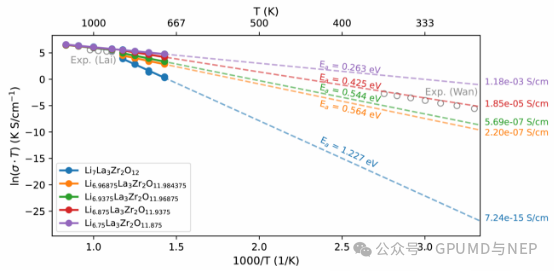
Figure 4. Arrhenius plot of lithium-ion diffusion in LLZO under different lithium nonstoichiometries. Gray open circles are the experimental results.
Point 3: The Main Driving Force for Increasing Ionic Conductivity Is Lithium Defects Rather Than Oxygen Defects
The main driving force for increasing the ionic conductivity of tetragonal LLZO is lithium defects rather than oxygen defects. Simulation results show that introducing only oxygen defects hardly changes the migration characteristics of lithium ions, and the activation energy remains above 1.2 eV with extremely low ionic conductivity. In contrast, after introducing lithium defects, regardless of the presence of oxygen defects, the activation energy of lithium ions is significantly reduced. This indicates that lithium defects play a key role in reducing the activation energy and increasing the ionic conductivity, while the main role of oxygen defects is to maintain the charge balance of the material.
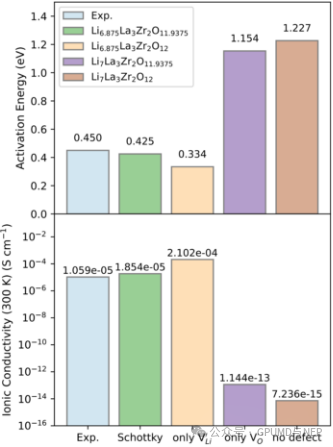
Figure 5. Activation energy and ionic conductivity of lithium ions in tetragonal LLZO with different defect types.
Point 4: Influence of Lithium Defects on Phase Transition Behavior
Lithium nonstoichiometry not only has an important impact on ion migration but also significantly regulates the tetragonal-cubic phase transition behavior. The research found that when the lithium defect concentration reaches about 3.6% (Li6.75), the tetragonal-cubic phase transition temperature is significantly reduced from 900 K to about 750 K. Similarly, the driving force for this phase transition mainly comes from lithium defects rather than the contribution of oxygen defects. The results show that by precisely controlling the lithium nonstoichiometry, the cubic phase with high conductivity can be stabilized at a lower temperature, providing new design ideas and strategies for optimizing the performance of LLZO electrolytes.
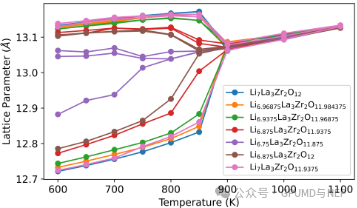
Figure 6. Evolution of the lattice parameters of intrinsic and nonstoichiometric LLZO.
Conclusions
This research is the first application of GPUMD&NEP in the field of solid-state electrolytes and successfully analyzes the impact of lithium nonstoichiometry on lithium-ion migration in LLZO. This breakthrough not only fills the long-term gap between theory and experiment but also demonstrates the broad application prospects of GPUMD&NEP in large-scale complex material systems.
The related results were published in the international well-known journal Chemistry of Materials under the title "Impact of Lithium Nonstoichiometry on Ionic Diffusion in Tetragonal Garnet-Type Li7La3Zr2O12 [1]". The first author is Dr. Yan Zihan, a doctoral student jointly cultivated by Zhejiang University and Westlake University, and Professor Zhu Yizhou is the sole corresponding author. All the calculations in this work were completed at the Supercomputing Center of Westlake University. The author revealed that for a system of 12288 atoms, a single 2080Ti can even achieve a calculation speed of about 211 timesteps/s, and a long-time simulation of 2 ns only took less than 3 hours.
Paper link: https://pubs.acs.org/doi/10.1021/acs.chemmater.4c02454
References
[1] Yan, Zihan, and Yizhou Zhu. "Impact of Lithium Nonstoichiometry on Ionic Diffusion in Tetragonal Garnet-Type Li7La3Zr2O12." Chemistry of Materials (2024).: https://pubs.acs.org/doi/10.1021/acs.chemmater.4c02454