What Can Uni-Mol Do Too? | Uni-Mol Docking Combined with BindingNet v2 Outperforms AlphaFold3 on Low-Similarity Molecules
In the field of drug research and development, accurately understanding the interactions between proteins and ligands is of great significance. It not only facilitates the discovery of new drugs but also provides a crucial basis for optimizing existing drugs. For a long time, the scarcity of high-quality data and the limitations of traditional research methods have greatly hindered the development of this field. Recently, researchers from the Institute of Biomedical Sciences at Tsinghua University and the Beijing Institute of Life Sciences have constructed the BindingNet v2 dataset. By combining it with the Uni-Mol model, they have made remarkable progress in structure-based drug design, especially in the task of predicting ligand binding sites of low-similarity molecules, where its performance exceeds that of AlphaFold3. The related research results were published in npj drug discovery under the title "Augmented BindingNet dataset for enhanced ligand binding pose predictions using deep learning". (doi: doi.org/10.1038/s44386 - 024 - 00003 - 0)
Research Background
Structure-based drug design relies on high-quality data of protein-ligand complex structures and binding affinities. However, existing datasets generally suffer from insufficient diversity and limited quantity, which severely restricts the comprehensive understanding of protein-ligand interactions. For example, the Protein Data Bank (PDB), although an important repository for biomolecular structures, has a negligible number of small molecules compared to the chemical space and lacks experimental binding affinity data for complex structures. Manually curated datasets such as Binding MOAD and PDBbind also have limitations and are difficult to meet the needs of drug research and development. Although many datasets based on molecular docking and template modeling, such as CrossDocked 2020 and BindingNet v1, have been constructed, they respectively have problems such as spatial conflicts between ligands and proteins and lack of updates. The expanded BindingNet v2 dataset has increased the scale and diversity of the data, providing higher value for the accuracy and reliability of deep learning models in predicting binding poses.
Method Introduction
Composition of the BindingNet v2 Dataset
The BindingNet v2, as a comprehensive and diverse dataset of protein-ligand complex structures, has the following remarkable features:
- Huge data scale: It contains 689,796 complex structures, covering 1,794 protein targets and 475,309 unique ligands, significantly expanding the scale of existing datasets.
- Innovative construction method: By combining pharmacophores and molecular shape similarity, it improves the modeling ability for low-similarity templates and solves the problem of dependence on high-similarity templates in traditional template modeling.
- Clear data classification: According to the model confidence level, the complexes are divided into three categories: high confidence (33.63%), medium confidence (23.91%), and low confidence (42.45%), providing a reliable reference standard for further research.
- Wide range of application scenarios: It supports tasks such as protein-ligand binding site prediction, binding affinity prediction, and molecular generation, providing rich data support for the development and optimization of deep learning models.
In addition, the BindingNet v2 dataset integrates data from multiple sources such as PDB and ChEMBL. Through strict screening and optimization, the quality of the data and its experimental relevance are ensured.

Figure 1 Information of the BindingNet v2 dataset. A. The type distribution of protein targets in the BindingNet v2 dataset, including kinases, enzymes, G protein-coupled receptors, etc.; B. The confidence classification of model structures in the dataset, divided into three categories: low, medium, and high; C. The distribution of experimental binding affinities. D. The stability analysis of binding affinity data.
Verification of the BindingNet v2 Dataset
To verify the practical effect of the BindingNet v2 dataset, the research team widely applied it in multiple deep learning models and focused on analyzing the performance of the models in the task of predicting ligand binding sites. The PoseBusters dataset was used as the test data for verification. The core indicators included the success rate of binding site prediction (defined as the proportion of ligands with RMSD < 2Å) and the PB-valid proportion after physical optimization (verifying the physical rationality of the binding site). This experimental design ensured a comprehensive evaluation of the prediction accuracy and scientific rationality of the models.
- Baseline performance of the PDBbind dataset: When training with the traditional PDBbind dataset, the Uni-Mol model's prediction performance for low-similarity molecules (Tanimoto coefficient Tc < 0.3) was relatively limited, with a success rate of only 38.55%. Although the model had a relatively high success rate for high-similarity molecules (Tc > 0.7), the problem of insufficient generalization ability was still obvious. This indicates that models relying on traditional datasets have difficulty effectively dealing with the challenges of structurally novel molecules in chemical space, further emphasizing the importance of larger-scale and more diverse datasets.
- Performance improvement of the BindingNet v2 dataset: After combining the BindingNet v2 dataset, the performance of the Uni-Mol model was significantly improved. In the prediction of low-similarity molecules, the success rate increased significantly from 38.55% to 64.25%, demonstrating the crucial role of dataset diversity in the model's generalization ability. In addition, after the research further introduced MM-GB/SA optimization and re-scoring methods, the success rate of Uni-Mol increased again to 74.07%, reaching a new height in the complex task of binding site prediction. These results fully demonstrate the unique value of the BindingNet v2 dataset in improving the performance of deep learning models.
- Verification results of other models: In comparison with other cutting-edge models, the Uni-Mol model combined with the BindingNet v2 dataset also performed outstandingly. In the drug molecule test of the PoseBusters dataset, the success rate of Uni-Mol reached 79.1%, significantly higher than 71.3% of AlphaFold3. This result shows that Uni-Mol has obvious advantages in the prediction ability for low-similarity molecules. In addition, the BindingNet v2 dataset also had a positive impact on the sampling and scoring abilities of traditional molecular docking methods (such as Glide), further verifying its wide applicability in multiple prediction methods.

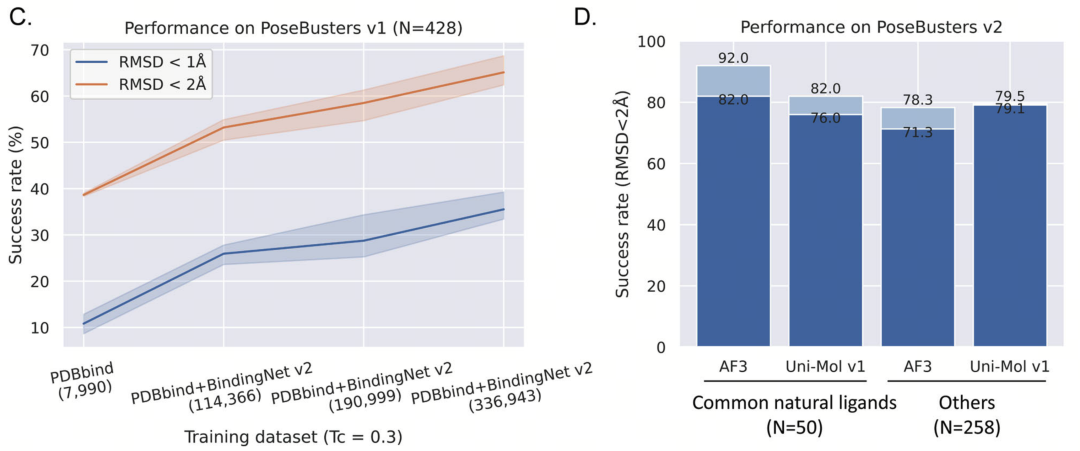
Figure 2 Results of training the Uni-Mol model using the BindingNet v2 dataset. A. It shows the similarity distribution (Tanimoto coefficient, Tc) of ligands in the PoseBusters v1 test set and ligands in the PDB (2019 version) dataset. Low-similarity ligands (Tc < 0.3) account for a large proportion; B. The prediction performance of the Uni-Mol model on training sets with different similarities (based on PDBbind v2020). The prediction success rate decreases as the similarity decreases; C. After introducing a larger dataset (such as BindingNet v2), the prediction success rate of Uni-Mol increases from 38.55% to 64.25%; D. The performance comparison between AlphaFold3 and Uni-Mol (combined with PDBbind + BindingNet v2). Uni-Mol performs better in binding site prediction and chemical rationality verification.
Conclusion
Research shows that the BindingNet v2 dataset, through its large scale, high diversity, and innovative construction method, provides rich training data for deep learning models and significantly improves the performance of protein-ligand binding site prediction. Especially in the application verification of the Uni-Mol model, the BindingNet v2 dataset not only improves the model's generalization ability for low-similarity molecules but also lays a foundation for its practical application in drug research and development scenarios.
However, the research also reveals some directions that need further optimization. For expanding the chemical and target space, in the future, it is possible to combine larger-scale virtual docking data and prediction structures such as AlphaFold to further enhance data diversity. Secondly, inactive data can be added to enhance the model's adaptability to virtual screening tasks. Finally, deeply integrating physics-based optimization tools with deep learning models can further improve the accuracy and physical rationality of prediction results.
The collaborative innovation of BindingNet v2 and the Uni-Mol model provides a powerful tool for the intelligent and efficient development of drugs. Future research is expected to play a greater potential in structure-based drug design and virtual screening.