What Can Uni-Mol Do too? | Full-Scale AI Design of Optoelectronic Materials from Molecules to Devices
From the vivid colors of smartphone displays and the high efficiency of photovoltaic solar panels, to high–energy-density batteries and sharp bio-fluorescent imaging, organic optoelectronic molecules are indispensable. They serve as the “soul” and “modulator” of optoelectronic functions. With structural tunability at the molecular scale, they continuously enable the evolution of optoelectronic devices and their broad application scenarios.
However, to fully unlock the potential of organic optoelectronic materials, it is crucial to efficiently understand—across multiple scales—the intrinsic links between molecular structure, material properties, and device performance.
Recently, the Functional Molecular Design Team of AI for Science Institute (AISI), together with the DP Technology development team, in collaboration with Peking University, Sinopec Research Institute of Petroleum Processing, Shandong University, Henan Normal University, Shenzhen Institute of Synthetic Biology, and several other institutions, introduced OCNet—a pretraining framework for organic optoelectronic materials built upon the Uni-Mol architecture. OCNet is trained on tens of millions of conjugated molecules and their dimers.
OCNet achieves, for the first time, a unified virtual representation spanning molecules, mesoscale materials, and devices: it surpasses existing SOTA models by 20% on molecular-scale performance, enables cross-material generalizable mobility prediction in amorphous organic thin films for the first time, and delivers near-real-time, high-accuracy prediction of device-level photovoltaic efficiency. The work has been published in npj Computational Materials (doi: 10.1038/s41524-025-01788-y).
Methodological Highlights
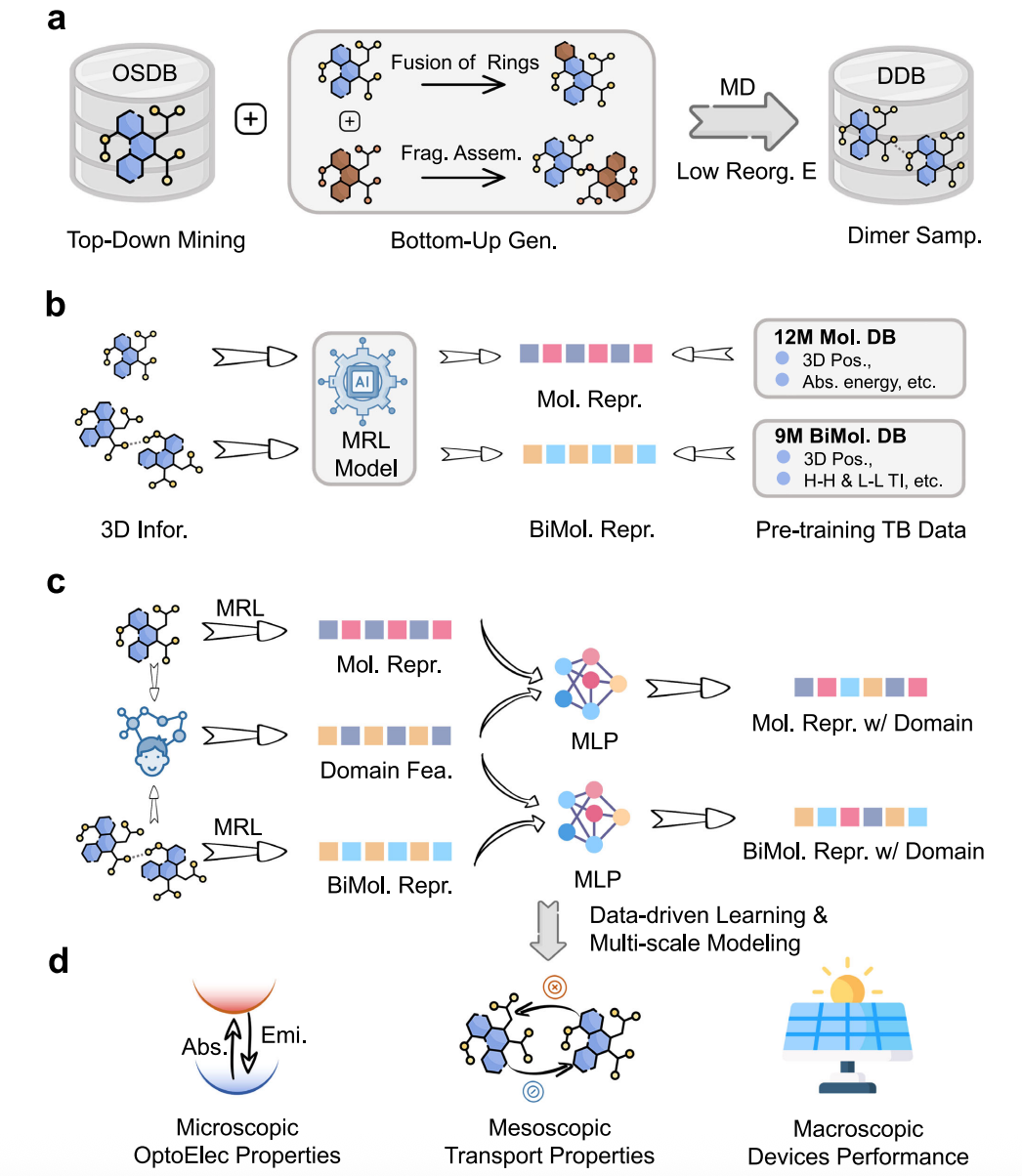
The OCNet framework (Fig. 1) builds on Uni-Mol and performs a second-stage pretraining to comprehensively capture the optoelectronic and charge-transport behavior of organic conjugated systems, starting from the molecular scale. Its core innovations include:
Tens-of-Millions-Scale Database
A database of over ten million molecules and dimers is constructed for the first time, covering metal–organic complexes, fused-ring systems, and fragment-assembled structures. A large-scale dimer configuration set under thin-film environments is also generated, dramatically expanding the covered chemical space.
Two-Stage Pretraining Strategy
OCNet first learns structural information, then learns optoelectronic properties and charge-transfer dynamics at the tight-binding level, thereby gaining deep physical knowledge.
Expert-Knowledge-Enhanced Learning
By integrating expert descriptors such as electronic-structure features, OCNet improves both physical interpretability and predictive accuracy.
Cross-Scale Representation & Prediction
From molecular excited-state properties, to thin-film charge mobility, to device-level power conversion efficiency (PCE), OCNet achieves unified modeling across scales.
With this new strategy, OCNet delivers a multi-scale virtual representation and performance prediction pipeline for organic functional materials—from molecules to full devices—achieving universality, accuracy, and efficiency.
Results and Breakthroughs
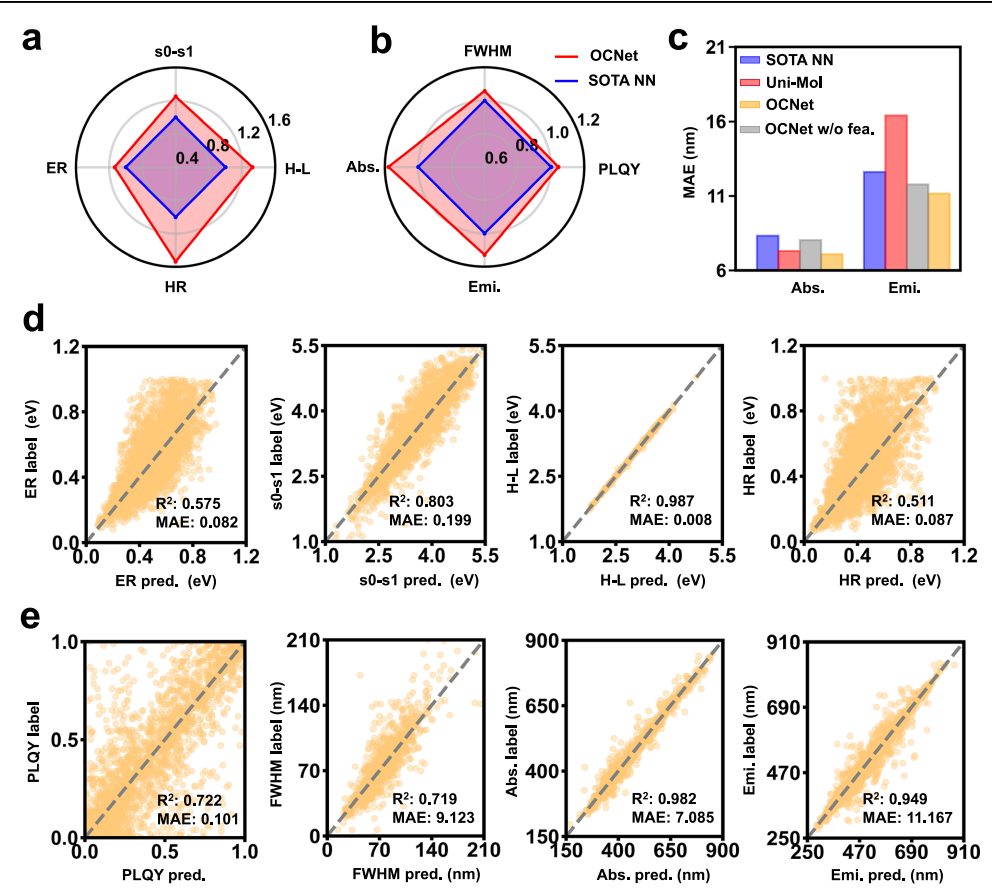
Molecular Scale: Large Performance Gains Beyond SOTA
As shown in Fig. 2, OCNet demonstrates breakthrough performance in predicting molecular optoelectronic properties. For both theoretical properties—such as HOMO–LUMO gaps, excitation energies, and reorganization energies—and experimental observables including absorption/emission wavelengths and quantum efficiency, OCNet significantly outperforms state-of-the-art models:
- Overall accuracy improves by more than 20%
- Certain tasks, such as reorganization-energy prediction, improve by up to 60%
This enables researchers to obtain quantum-chemistry-level precision at much lower computational cost, providing strong support for large-scale candidate material screening.
Mesoscale: First-Ever Thin-Film Mobility Prediction, Highly Consistent with DFT
On the mesoscale (Fig. 3a, b), OCNet achieves thin-film charge-transfer integral prediction and, through multi-scale modeling, accurately reconstructs charge mobility in organic semiconductors.
Compared with DFT benchmarks:
- OCNet outputs show excellent agreement
- The correlation coefficient R reaches 0.94 for log electron mobility
- OCNet dramatically surpasses approximate tight-binding approaches (e.g., GFN-xTB), which suffer from systematic underestimation
This fills the long-standing gap in thin-film mobility prediction and lays the groundwork for high-throughput discovery of high-mobility organic semiconductors.
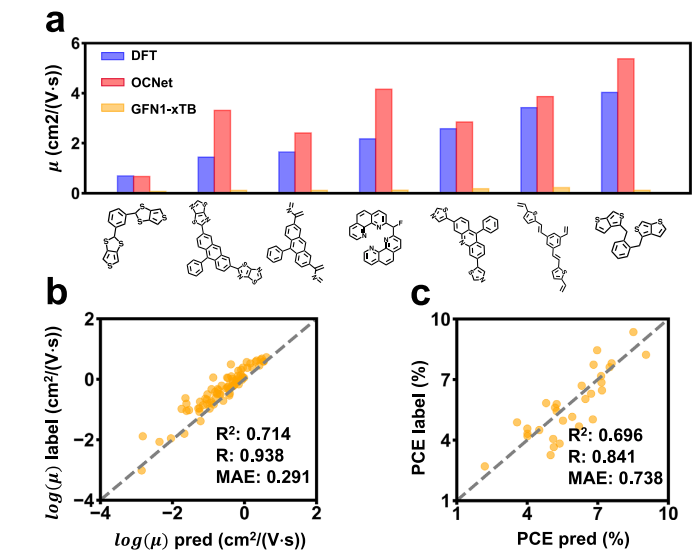
Device Scale: Near-Real-Time, High-Accuracy PCE Prediction
At the device level (Fig. 3c), OCNet delivers near-real-time prediction of organic photovoltaic device power conversion efficiency (PCE). Leveraging low-cost tight-binding electronic-structure features:
- Correlation coefficient R reaches 0.84
- Accuracy significantly surpasses models based solely on electronic-structure descriptors
- Runtime per prediction: 0.005 seconds
- In contrast, generating TDDFT-based descriptors requires thousands of CPU hours
This combination of high precision and high efficiency makes OCNet a truly practical tool for virtual device-performance characterization.
Future Outlook
OCNet breaks the traditional bottlenecks of optoelectronic materials research by achieving, for the first time, cross-scale virtual representations from molecules to devices—substantially improving both efficiency and accuracy in performance prediction.
In the future, OCNet will further integrate high-throughput experiments to form a closed loop of virtual characterization → experimental validation → intelligent iteration, accelerating the discovery and deployment of new materials.
More importantly, OCNet may accelerate patent and intellectual-property strategies across molecular databases, cross-scale modeling, and device-performance prediction, forming new technological barriers and enabling rapid transfer of research advances to industry.
This direction aligns strongly with China’s “AI+” national strategy. With supportive policies in AI-for-materials and AI-for-energy, OCNet will empower strategic emerging industries—including photovoltaics, displays, and sensing—and help drive independent innovation and global leadership in intelligent optoelectronics and materials design.
The Uni-Mol architecture demonstrates exceptional adaptability and domain application depth in this work. This is the first successful application of Uni-Mol to full-scale modeling of optoelectronic materials, establishing a new AI paradigm for designing organic optoelectronic molecules and accelerating the development of next-generation functional materials.
Paper: https://www.nature.com/articles/s41524-025-01788-y
Code, model weights & data: https://github.com/545487677/OCNet