What Can Uni-Mol Do Too? | Active Molecule Search from Large-Scale Molecular Libraries without Protein Structures
Virtual screening is a crucial technique in the early stage of drug discovery, aiming to identify potential drug candidate molecules from vast molecular libraries. Ligand - based virtual screening, such as molecule search, has drawn significant attention as it does not rely on specific protein site information. Recently, researchers Zhen Wang, Feng Yu, Guolin Ke, and Zhifeng Gao from DP Technology, in collaboration with Professor Zhewei Wei from Renmin University of China and doctoral student Gengmo Zhou, published a paper titled "S - MolSearch: 3D Semi - supervised Contrastive Learning for Bioactive Molecule Search" at the top machine learning conference NeurIPS 2024. This paper introduced in detail a 3D semi - supervised learning framework called S - MolSearch for active molecule search. The S - MolSearch is designed based on the principle of inverse optimal transport and can effectively combine and utilize labeled and unlabeled data, demonstrating a remarkable improvement over existing virtual screening methods. Uni - Mol functions as a 3D molecule encoder and plays a central role in this process, showcasing its great potential in molecule representation and molecule similarity measurement and providing strong support for new drug discovery.
Research Background
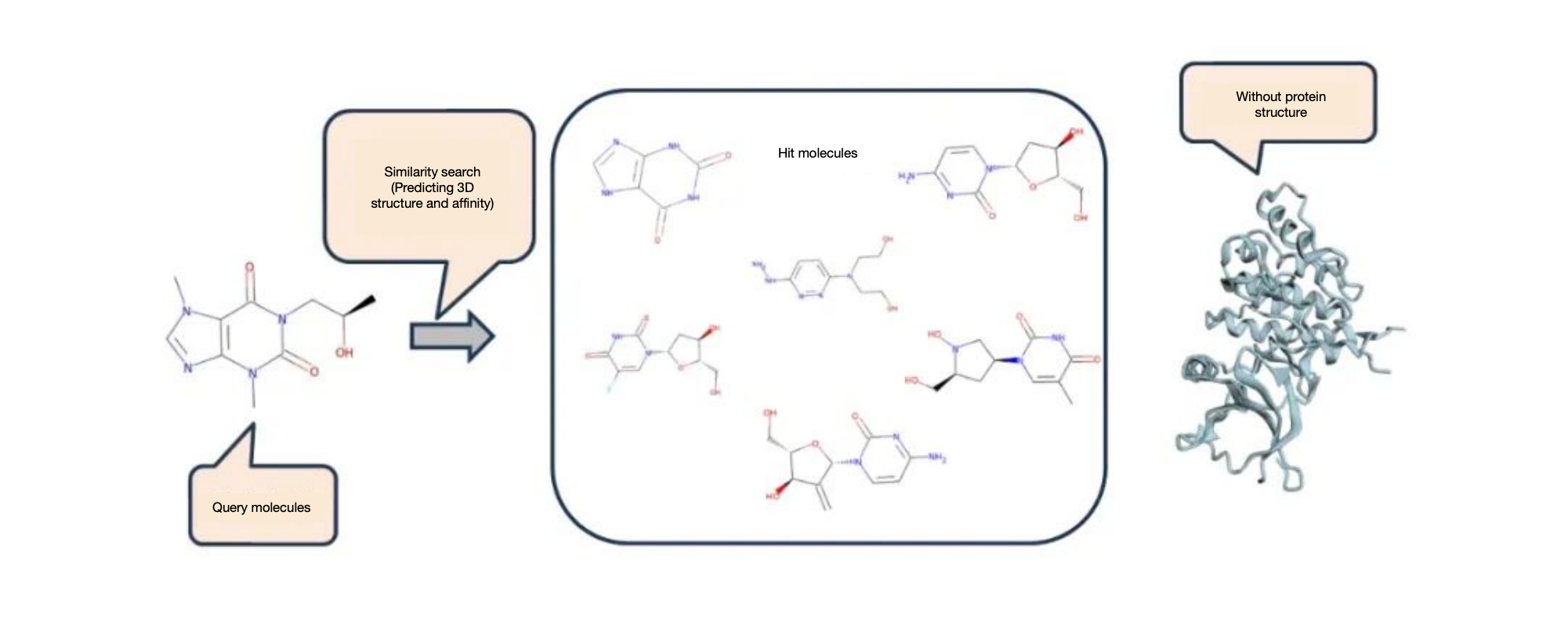
Molecule search is one of the core techniques in ligand-based virtual screening. Traditional molecule search methods usually focus on the structural features of molecules, such as molecular fingerprints and conformations, to measure the similarity between molecules. However, most of these methods fail to fully consider the similarity in biological activity of molecules. Screening only based on structural features may lead to missing many potentially active molecules, thus reducing the accuracy and efficiency of virtual screening.
One approach is to directly use the binding affinity data of protein-ligand complexes. However, the acquisition process of these data is often expensive and time-consuming, resulting in limited available labeled data, which only covers a small part of the chemical space. At the same time, many unlabeled molecular data are relatively easy to obtain. Although they do not have clear biological activity annotations, they can reflect the chemical characteristics and structural diversity of molecules to a certain extent, cover a broader chemical space, and can provide richer training information for the model. A natural idea is how to effectively combine labeled and unlabeled data and fully exploit their potential to improve the accuracy of molecule search.
In this paper, by designing an appropriate semi-supervised learning method S-MolSearch, the labeled data and unlabeled data can be effectively combined and utilized to improve the performance and generalization ability of molecule search.
Method Introduction
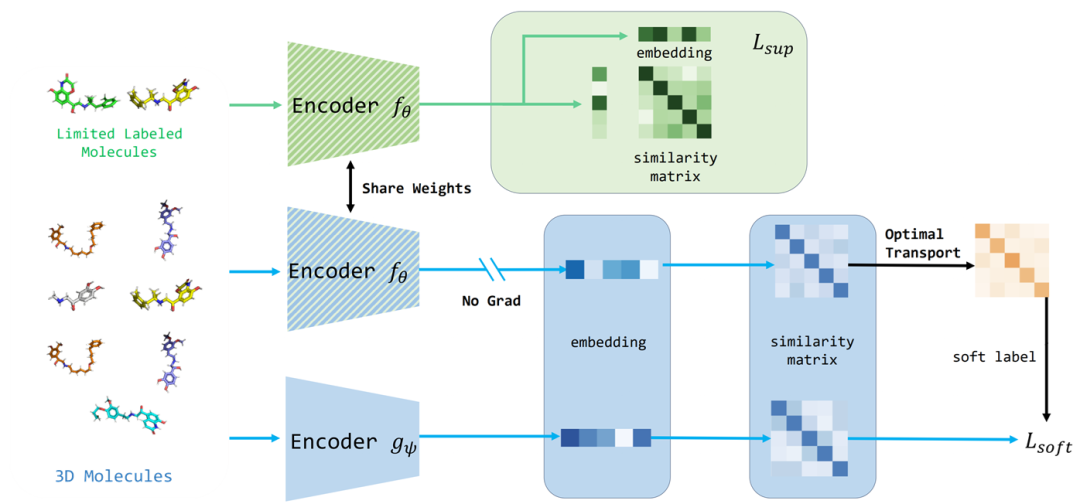
The architecture diagram of S-MolSearch. The green part on the top is the process of the encoder fθ being trained with affinity data, while the blue part at the bottom is the process of the encoder gψ being trained with the whole data and soft labels.
To address the above challenges, this paper proposes S-MolSearch, a new 3D semi-supervised contrastive learning framework. The S-MolSearch uses 3D molecular structures to capture structural similarity and combines affinity data and unlabeled data in a semi-supervised manner to capture biological activity similarity.
The S-MolSearch consists of two main components:
- Encoder trained with labeled dataset fθ: The authors constructed a labeled molecule-protein binding dataset from ChEMBL. According to the binding affinity, molecules are assigned to different targets, and the active molecules corresponding to each target form a cluster. Then, samples are taken from these classes. Molecules that can bind to the same target are regarded as positive samples, and those that bind to different targets are regarded as negative samples. This supervised contrastive learning is used to introduce affinity information and avoid the limitation of only relying on structural similarity for search.
- Encoder trained with the whole dataset gψ: Here, the whole data refers to the molecular data in the labeled data and the unlabeled molecular data. The authors assume that the similarity measure obtained by the encoder fθ can be generalized to unlabeled data. During training, the same data from the whole dataset is input into these two encoders respectively, and soft labels are obtained from fθ using the optimal transport method. Then, these soft labels are used to train the encoder gψ. This method enables S-MolSearch to effectively utilize unlabeled data and ensures that the model can learn from both the affinity-labeled samples and the broader structural similarity in the molecular dataset.
In this paper, Uni-Mol is selected as the 3D molecule encoder. The encoder fθ is used to process the "Limited Labeled Molecules". After passing through the encoder fθ, it will generate an "embedding" and a "similarity matrix", which is related to Lsup. The encoder gψ is used to process the "3D Molecules". After going through the encoder gψ, it will obtain an "embedding" and then derive a "similarity matrix", which is associated with Lsoft. It helps extract molecule representations and rank the molecules in the library according to their similarity with the query molecule representation, so as to identify candidate molecules.
Results and Discussion
During the training, the whole data used by the authors comes from the pre-trained data of Uni-Mol and the molecular data in ChEMBL, with a total of approximately 19 million. The labeled data comes from ChEMBL, about 1 million.
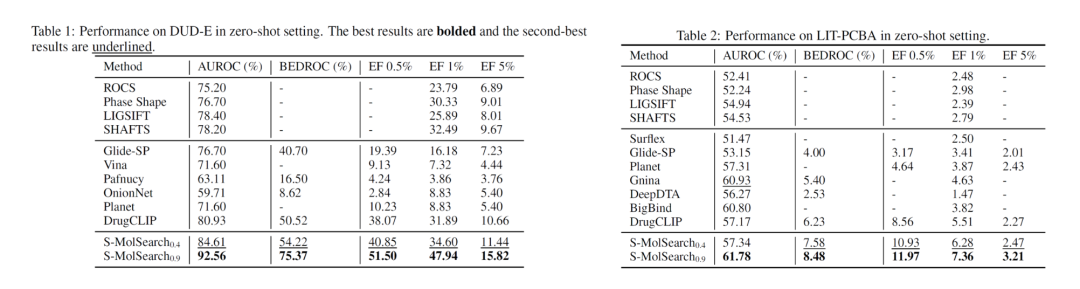
In the experiment, S-MolSearch performs excellently on the benchmark datasets LIT-PCBA and DUD-E widely used in virtual screening. It outperforms existing structure-based virtual screening methods and label-based virtual screening methods in terms of AUROC, BEDROC, and EF metrics. These methods include traditional 3D similarity search tools, docking software, and deep learning-based methods.
Here, the authors trained two models according to different similarity thresholds of proteins in the training set and test set. The subscript of the threshold indicates that the data with a protein sequence similarity higher than the threshold in the test set is removed from the training set. The 0.4 version model avoids overfitting on similar targets, while the 0.9 version model is significantly better than the best baseline. For example, on the DUD-E data, the improvement of BEDROC exceeds 49%, and the improvement of EF exceeds 30%.
The authors also conducted ablation studies to compare the effects of different modules, such as whether to use soft labels, whether to add regularization losses, and whether to use pre-trained models. The results show that each part has a positive contribution to the results.
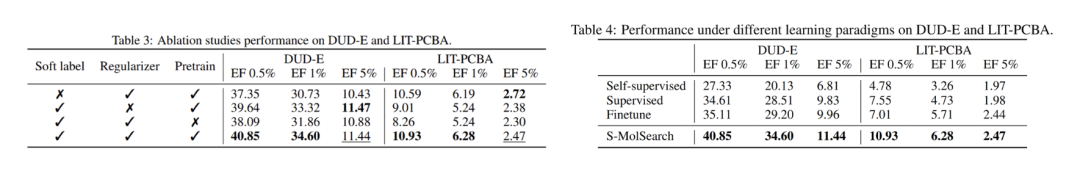
Subsequently, the authors also studied different training methods using unlabeled data and labeled data. Under the premise of contrastive learning, the semi-supervised learning method performs better than self-supervised, supervised, or fine-tuning methods.
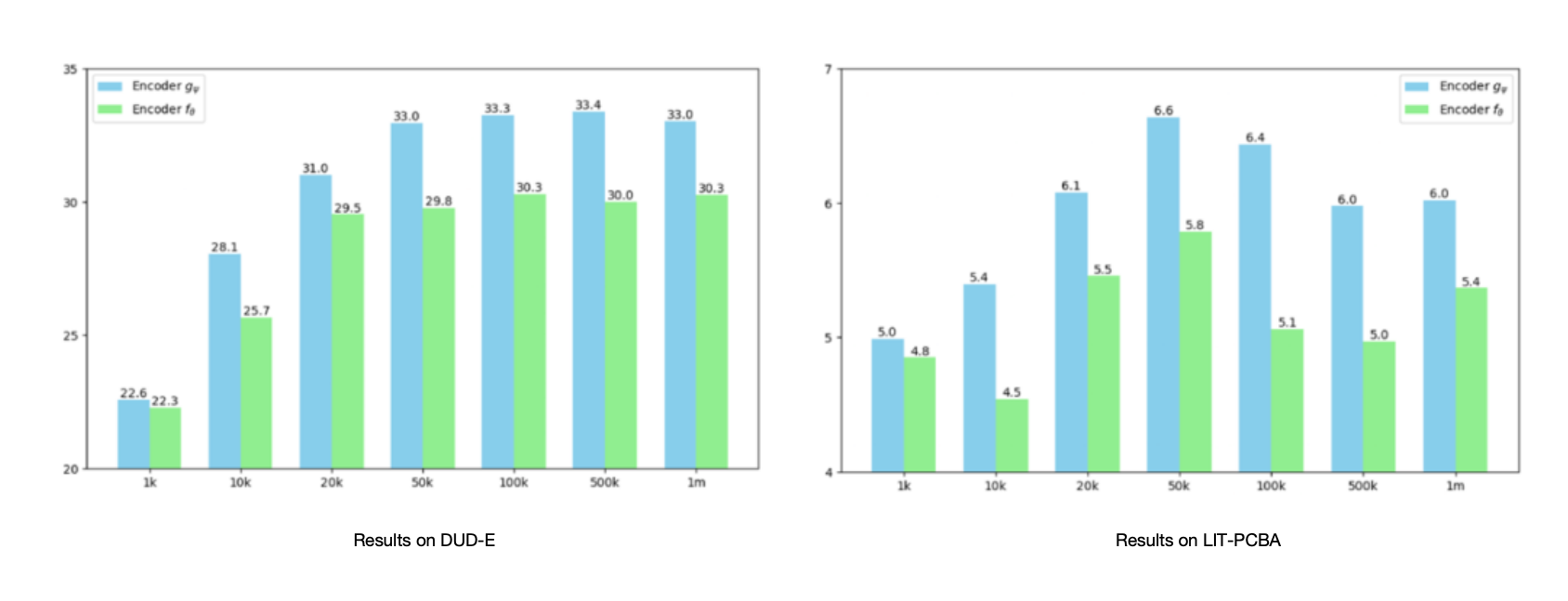
Since the acquisition cost of labeled data is high in the field of drug discovery, the authors also analyzed how the scale of labeled data affects the results in semi-supervised learning. As shown in the figure, the experiment was carried out with different amounts of labeled data while keeping the unlabeled data fixed at 1 million. With the increase of the amount of labeled data, the performance of the encoder gψ improves, especially when the amount of labeled data is limited. Moreover, the results of the encoder gψ are always higher than those of the encoder fθ trained only with labeled data. The best results appear when the labeled data is 50,000 and 100,000, corresponding to the labeled and unlabeled data ratios of 1:20 and 1:10. After further increasing the labeled data, the model performance remains stable or slightly decreases, indicating that further improvement may require increasing unlabeled data or re-searching for parameters.
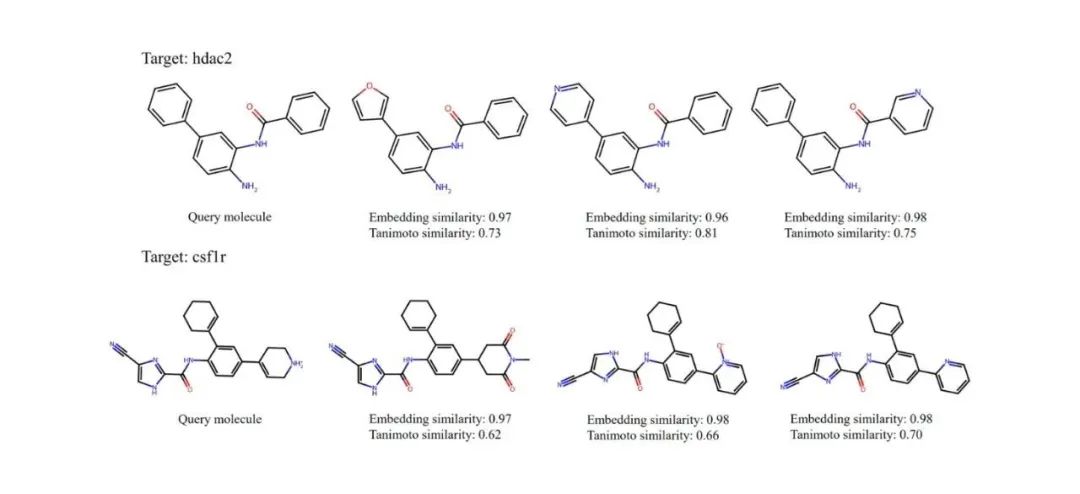
The authors also provided qualitative examples to help understand the capabilities of S-MolSearch. They selected two targets, hdac2 and csf1r, from the DUD-E dataset. As shown in the figure, the query molecule and the top-ranked molecules retrieved by S-MolSearch are displayed. These molecules are all active molecules, and the embedding similarity and Tanimoto similarity of the molecular fingerprints between these molecules and the query molecule are given. The results show that molecules with higher fingerprint similarity usually also have higher embedding similarity.
Conclusion
This study introduced S-MolSearch, a new semi-supervised contrastive learning framework, which significantly enhanced the generalization ability of machine learning models in virtual screening. Based on the inverse optimal transport, S-MolSearch ingeniously combines limited labeled data with a large amount of unlabeled data to help identify potential drug candidate molecules from large-scale molecular libraries and significantly improve the accuracy and efficiency of molecule search. Uni-Mol plays an important role as a 3D molecule encoder, integrating molecular structure information into the screening framework.
Currently, S-MolSearch mainly focuses on molecular affinity data and does not involve broader biochemical interactions, which provides a potential direction for further improvement. Future work can integrate more unsupervised data to further enhance the effectiveness of the framework and explore its more applications in different bioinformatics fields.
Bohrium App Address
Click the following link to jump to the Bohrium® App for trial: https://bohrium.dp.tech/apps/s-molsearch