The following publications have used the ABACUS software. Publications that only mentioned the ABACUS will not be included below.
We encourage explicitly mentioning ABACUS with proper citations in your publications, so we can more easily find and list these publications.
Last update date: June 18, 2025
2025
Tuning oxygen vacancies in complex oxides using 2D layered materials
Jiangbo Luo, Xudong Zhu, Xu Lian, Yuntian Zheng, Reshmi Thottathil, Wei Chen, Song Liu, A Ariando, Junxiong Hu
2D Mater., 2025, 12 (1), 15022.
DOI: 10.1088/2053-1583/ada041
Interlayer coupling driven rotation of the magnetic easy axis in MnSe2 monolayers and bilayers
Zhongqin Zhang, Cong Wang, Peng-Jie Guo, Linwei Zhou, Yuhao Pan, Zhixin Hu, Wei Ji
Physical Review B, 2025, 111, 054422.
DOI: 10.1103/PhysRevB.111.054422

Interfacial polarization-induced tribological behavior in MoS2/β-Te and G/β-Te heterostructures
Guoliang Ru, Weihong Qi, Kaiyuan Xue, Mengzhao Wang, Xuqing Liu
Nanoscale, 2025, 17, 7497–7509.
DOI: 10.1039/D4NR05628F
Interlaced nanotwinned diamond and its deformation mechanism under pure shear strain
Mingqiang Zhang, Yabei Wu, Ye Sheng, Jing Huang, Yanxiao Hu, Xiaoxin Xu, Xuezhi Ke, Wenqing Zhang
Materials Today Physics, 2025, 52, 101685.
DOI: 10.1016/j.mtphys.2025.101685
Mechanistic insights into temperature effects for ionic conductivity in Li6PS5Cl
Zicun Li, Jianxing Huang, Xinguo Ren, Jinbin Li, Ruijuan Xiao, Hong Li
Journal of Power Sources, 2025, 640, 236632.
DOI: 10.1016/j.jpowsour.2025.236632
Hybrid gauge approach for accurate real-time TDDFT simulations with numerical atomic orbitals
Haotian Zhao, Lixin He
Journal of Chemical Theory and Computation, 2025, 21, 7–18.
DOI: 10.1021/acs.jctc.5c00111
Efficient hybrid-functional-based force and stress calculations for periodic systems with thousands of atoms
Peize Lin, Yuyang Ji, Lixin He, Xinguo Ren
Journal of Chemical Theory and Computation, 2025, 21, 3394–3407.
DOI: 10.1021/acs.jctc.4c01635
Direct minimization on the complex Stiefel manifold in Kohn--Sham density functional theory for finite and extended systems
Kai Luo, Tingguang Wang, Xinguo Ren
Computer Physics Communications, 2025, 312, 109596.
DOI: 10.1016/j.cpc.2025.109596
DeePMD-GNN: A DeePMD-kit plugin for external graph neural network potentials
Jinzhe Zeng, Timothy J. Giese, Duo Zhang, Han Wang, Darrin M. York
Journal of Chemical Information and Modeling, 2025, 65, 3154–3166.
DOI: 10.1021/acs.jcim.4c02441
Semimetallic 2D defective graphene networks with periodic 4--8 defect lines
Roland Gillen, Janina Maultzsch
physica status solidi (b), 2025, 259, 2500009.
DOI: 10.1002/pssb.202500009
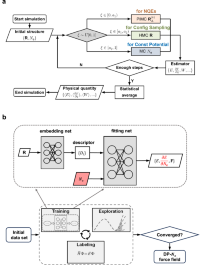
Probing nuclear quantum effects in electrocatalysis via a machine-learning enhanced grand canonical constant potential approach
Menglin Sun, Bin Jin, Xiaolong Yang, Shenzhen Xu
Nature Communications, 2025, 16, 3600.
DOI: 10.1038/s41467-025-58871-7
Electric-field-independent spin-orbit-coupling gap in h-BN-encapsulated bilayer graphene
Fang-Ming Jing, Zhen-Xiong Shen, Guo-Quan Qin, Wei-Kang Zhang, Ting Lin, Ranran Cai, Zhuo-Zhi Zhang, Gang Cao, Lixin He, Xiang-Xiang Song, Guo-Ping Guo
Physical Review Applied, 2025, 23, 044053.
DOI: 10.1103/PhysRevApplied.23.044053
First-principles calculations and theoretical analysis of $\pi$ plasmon in graphene and graphite: From 2D to 3D
Pengfei Li, Ningju Hui
Vacuum, 2025, 240, 114424.
DOI: 10.1016/j.vacuum.2025.114424
Fundamentals of plane wave-based methods for energy band calculations in solids
Shengxin Yang, Kan-Hao Xue, Xiangshui Miao
Journal of Physics: Condensed Matter, 2025, 37, 233001.
DOI: 10.1088/1361-648X/adcf6b
FT2DP: Large atomic model fine-tuned machine learning potential for accelerating atomistic simulation of iron-based Fischer--Tropsch synthesis
Zhao-Qing Liu, Zhe Deng, Huabo Zhao, Han Wang, Mohan Chen, Hong Jiang
Journal of Materials Informatics, 2025, 5, 27.
DOI: 10.20517/jmi.2024.105
A deep learning framework for the electronic structure of water: Toward a universal model
Xinyuan Liang, Renxi Liu, Mohan Chen
Journal of Chemical Theory and Computation, 2025, 21, 14–27.
DOI: 10.1021/acs.jctc.5c00496
Modulation of electric field and interface on competitive reaction mechanisms
Pengchao Zhang, Xuefei Xu
Journal of Chemical Theory and Computation, 2025, 21, 6584–6596.
DOI: 10.1021/acs.jctc.5c00705
Suppression of irradiation defects and crack propagation in niobium via grain boundary engineering: A deep potential molecular dynamics study
Jiahang Li, Yajun Zhang, Jie Wang, Huadong Yong
Materials \& Design, 2025, 256, 114292.
DOI: 10.1016/j.matdes.2025.114292
Deep variational free energy prediction of dense hydrogen solid at 1200 K
Xinyang Dong, Hao Xie, Yixiao Chen, Wenshuo Liang, Linfeng Zhang, Lei Wang, Han Wang
Physical Review B, 2025, 111, 214118.
DOI: 10.1103/rbsg-r7hx
First-principles prediction of shock Hugoniot curves of boron, aluminum, and silicon from stochastic density functional theory
Tao Chen, Qianrui Liu, Chang Gao, Mohan Chen
Matter and Radiation at Extremes, 2025, 10, 057601.
DOI: 10.1063/5.0224902
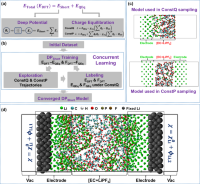
Observation of dendrite formation at Li metal--electrolyte interface by a machine-learning enhanced constant potential framework
Taiping Hu, Haichao Huang, Guobing Zhou, Xinyan Wang, Jiaxin Zhu, Zheng Cheng, Fangjia Fu, Xiaoxu Wang, Fuzhi Dai, Kuang Yu, Shenzhen Xu
Nature Communications, 2025, 16, 7379.
DOI: 10.1038/s41467-025-62824-5
Synergistic mechanisms in SnO2@graphene for ambient temperature NH3 sensing: A DFT study
Bo Liu, Xu Tang, Xinting Su
Chemical Engineering Journal, 2025, 521, 167087.
DOI: 10.1016/j.cej.2025.167087
2024
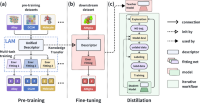
DPA-2: a large atomic model as a multi-task learner
Duo Zhang, Xinzijian Liu, Xiangyu Zhang, Chengqian Zhang, Chun Cai, Hangrui Bi, Yiming Du, Xuejian Qin, Anyang Peng, Jiameng Huang, Bowen Li, Yifan Shan, Jinzhe Zeng, Yuzhi Zhang, Siyuan Liu, Yifan Li, Junhan Chang, Xinyan Wang, Shuo Zhou, Jianchuan Liu, Xiaoshan Luo, Zhenyu Wang, Wanrun Jiang, Jing Wu, Yudi Yang, Jiyuan Yang, Manyi Yang, Fu-Qiang Gong, Linshuang Zhang, Mengchao Shi, Fu-Zhi Dai, Darrin M. York, Shi Liu, Tong Zhu, Zhicheng Zhong, Jian Lv, Jun Cheng, Weile Jia, Mohan Chen, Guolin Ke, Weinan E, Linfeng Zhang, Han Wang
npj Comput. Mater, 2024, 10 (1), 293.
DOI: 10.1038/s41524-024-01493-2
Uniformity, Linearity, and Symmetry Enhancement in TiOx/MoS2-xOx Based Analog RRAM via S-Vacancy Confined Nanofilament
Dongdong Sun, Xudong Zhu, Shaochuan Chen, Haotian Fang, Guixu Zhu, Gongpeng Lan, Lixin He, Yuanyuan Shi
Nano Lett., 2024, 24 (51), 16283–16292.
DOI: 10.1021/acs.nanolett.4c04434
Short-range order and strong interplay between local and itinerant magnetism in GeFe3N
Tinghai Zhang, Yantao Cao, Bo Zhang, Hanjie Guo, Liang Qiao, Fashen Li, Zhiwei Li
Phys. Rev. B, 2024, 110 (22), 224419.
DOI: 10.1103/PhysRevB.110.224419
Evolution of flat bands in MoSe2/WSe2 moir'e lattices: A study combining machine learning and band unfolding methods
Shengguo Yang, Jiaxin Chen, Chao-Fei Liu, Mingxing Chen
Phys. Rev. B, 2024, 110 (23), 235410.
DOI: 10.1103/PhysRevB.110.235410
Multi-channel machine learning based nonlocal kinetic energy density functional for semiconductors
Liang Sun, Mohan Chen
Electron. Struct., 2024, 6 (4), 45006.
DOI: 10.1088/2516-1075/ad8b8c
Substantially Enhanced Spin Polarization in Epitaxial CrTe2 Quantum Films
Xiaoqian Zhang, Qiangsheng Lu, Zhen-Xiong Shen, Wei Niu, Xiangrui Liu, Jiahua Lu, Wenting Lin, Lulu Han, Yakui Weng, Tianhao Shao, Pengfei Yan, Quan Ren, Huayao Li, Tay- Rong Chang, David J. Singh, Lixin He, Liang He, Chang Liu, Guang Bian, Lin Miao, Yongbing Xu
Adv. Mater. (Deerfield Beach Fla,), 2024, e2411137.
DOI: 10.1002/adma.202411137
Investigating interfacial segregation of $\Omega$/Al in Al-Cu alloys: A comprehensive study using density functional theory and machine learning
Yu Liu, Yin Zhang, Namin Xiao, Xingwu Li, Fu-Zhi Dai, Mohan Chen
Acta Mater., 2024, 279, 120294.
DOI: 10.1016/j.actamat.2024.120294
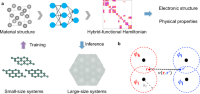
A deep equivariant neural network approach for efficient hybrid density functional calculations
Zechen Tang, He Li, Peize Lin, Xiaoxun Gong, Gan Jin, Lixin He, Hong Jiang, Xinguo Ren, Wenhui Duan, Yong Xu
Nat. Commun., 2024, 15 (1), 8815.
DOI: 10.1038/s41467-024-53028-4
Accelerating the calculation of electron-phonon coupling strength with machine learning
Yang Zhong, Shixu Liu, Binhua Zhang, Zhiguo Tao, Yuting Sun, Weibin Chu, Xin-Gao Gong, Ji-Hui Yang, Hongjun Xiang
Nat. Comput. Sci., 2024, 4 (8), 615–625.
DOI: 10.1038/s43588-024-00668-7
Deep learning tight-binding approach for large-scale electronic simulations at finite temperatures with ab initio accuracy
Qiangqiang Gu, Zhanghao Zhouyin, Shishir Kumar Pandey, Peng Zhang, Linfeng Zhang, Weinan E
Nat. Commun., 2024, 15 (1), 6772.
DOI: 10.1038/s41467-024-51006-4
Nonlocal free-energy density functional for a broad range of warm dense matter simulations
Cheng Ma, Min Chen, Yu Xie, Qiang Xu, Wenhui Mi, Yanchao Wang, Yanming Ma
Phys. Rev. B, 2024, 110 (8), 85113.
DOI: 10.1103/PhysRevB.110.085113
Geometric and electronic structures of Cs2BB'X6 double perovskites: The importance of exact exchange
Yuyang Ji, Peize Lin, Xinguo Ren, Lixin He
Phys, Rev, Res., 2024, 6 (3), 33172.
DOI: 10.1103/PhysRevResearch.6.033172
Effects of nonlocal pseudopotentials on the electrical and thermal transport properties of aluminum: A density functional theory study
Qianrui Liu, Mohan Chen
Phys. Rev. B, 2024, 110 (1), 14207.
DOI: 10.1103/PhysRevB.110.014207
Machine-Learning-Based Interatomic Potentials for Group IIB to VIA Semiconductors: Toward a Universal Model
Jianchuan Liu, Xingchen Zhang, Tao Chen, Yuzhi Zhang, Duo Zhang, Linfeng Zhang, Mohan Chen
J. Chem. Theory Comput., 2024, 20 (13), 5717–5731.
DOI: 10.1021/acs.jctc.3c01320
Concentrated solar CO2 reduction in H2O vapour with $>$1% energy conversion efficiency
Yuqi Ren, Yiwei Fu, Naixu Li, Changjun You, Jie Huang, Kai Huang, Zhenkun Sun, Jiancheng Zhou, Yitao Si, Yuanhao Zhu, Wenshuai Chen, Lunbo Duan, Maochang Liu
Nat. Commun., 2024, 15 (1), 4675.
DOI: 10.1038/s41467-024-49003-8
Family behavior and Dirac bands in armchair nanoribbons with 4-8 defect lines
Roland Gillen, Janina Maultzsch
J. Phys., Condens. Matter: Inst. Phys. J., 2024, 36 (29), 295501.
DOI: 10.1088/1361-648X/ad3b5a
Intramolecular and Water Mediated Tautomerism of Solvated Glycine
Pengchao Zhang, Axel Tosello Gardini, Xuefei Xu, Michele Parrinello
J. Chem. Inf. Model., 2024, 64 (9), 3599–3604.
DOI: 10.1021/acs.jcim.4c00273
Efficient Homojunction Tin Perovskite Solar Cells Enabled by Gradient Germanium Doping
Zhenzhu Zhao, Mulin Sun, Yuyang Ji, Kaitian Mao, Zongming Huang, Chengjian Yuan, Yuqian Yang, Honghe Ding, Yingguo Yang, Yu Li, Wenjing Chen, Junfa Zhu, Jing Wei, Jixian Xu, Watcharaphol Paritmongkol, Antonio Abate, Zhengguo Xiao, Lixin He, Qin Hu
Nano Lett., 2024, 24 (18), 5513–5520.
DOI: 10.1021/acs.nanolett.4c00646
Tuning of Berry-curvature dipole in TaAs slabs: An effective route to enhance the nonlinear Hall response
Hongsheng Pang, Gan Jin, Lixin He
Phys, Rev, Mater., 2024, 8 (4), 43403.
DOI: 10.1103/PhysRevMaterials.8.043403
Machine learning based nonlocal kinetic energy density functional for simple metals and alloys
Liang Sun, Mohan Chen
Phys. Rev. B, 2024, 109 (11), 115135.
DOI: 10.1103/PhysRevB.109.115135
Integration of Co Single Atoms and Ni Clusters on Defect-Rich ZrO2 for Strong Photothermal Coupling Boosts Photocatalytic CO2 Reduction
Jinghang Chen, Yuqi Ren, Yiwei Fu, Yitao Si, Jie Huang, Jiancheng Zhou, Maochang Liu, Lunbo Duan, Naixu Li
Acs Nano, 2024, 18 (20), 13035–13048.
DOI: 10.1021/acsnano.4c01637
Investigating structural and electronic properties of neutral zinc clusters: a G0W0 and G0W0\CYRG0(1) benchmark
Sunila Bakhsh, Muhammad Khalid, Sameen Aslam, Muhammad Sohail, Muhammad Aamir Iqbal, Mujtaba Ikram, Kareem Morsy
Beilstein J. Nanotechnol., 2024, 15, 310–316.
DOI: 10.3762/bjnano.15.28
Tuning flat bands by interlayer interaction, spin-orbital coupling, and external fields in twisted homotrilayer MoS2
Yonggang Li, Zhen Zhan, Shengjun Yuan
Phys. Rev. B, 2024, 109 (8), 85118.
DOI: 10.1103/PhysRevB.109.085118
Peculiar band geometry induced giant shift current in ferroelectric SnTe monolayer
Gan Jin, Lixin He
npj Comput. Mater, 2024, 10 (1), 23.
DOI: 10.1038/s41524-024-01213-w
Ab initio electronic structure calculations based on numerical atomic orbitals: Basic fomalisms and recent progresses
Peize Lin, Xinguo Ren, Xiaohui Liu, Lixin He
Wires Comput. Mol Sci, 2024, 14 (1).
DOI: 10.1002/wcms.1687
Ultrafast shift current dynamics in WS2 monolayer
Fuxiang He, Daqiang Chen, Xinguo Ren, Sheng Meng, Lixin He
Phys, Rev, Res., 2024, 6 (1), 13123.
DOI: 10.1103/PhysRevResearch.6.013123
Subquadratic-scaling real-space random phase approximation correlation energy calculations for periodic systems with numerical atomic orbitals
Rong Shi, Peize Lin, Min-Ye Zhang, Lixin He, Xinguo Ren
Phys. Rev. B, 2024, 109 (3), 35103.
DOI: 10.1103/PhysRevB.109.035103
Combining stochastic density functional theory with deep potential molecular dynamics to study warm dense matter
Tao Chen, Qianrui Liu, Yu Liu, Liang Sun, Mohan Chen
Matter Radiat. Extrem., 2024, 9 (1).
DOI: 10.1063/5.0163303
2023
Universal interatomic potential for perovskite oxides
Jing Wu, Jiyuan Yang, Yuan-Jinsheng Liu, Duo Zhang, Yudi Yang, Yuzhi Zhang, Linfeng Zhang, Shi Liu
Phys. Rev. B, 2023, 108 (18), L180104.
DOI: 10.1103/PhysRevB.108.L180104
PYATB: An efficient Python package for electronic structure calculations using ab initio tight-binding model
Gan Jin, Hongsheng Pang, Yuyang Ji, Zujian Dai, Lixin He
Comput. Phys. Commun., 2023, 291, 108844.
DOI: 10.1016/j.cpc.2023.108844
DeePKS Model for Halide Perovskites with the Accuracy of a Hybrid Functional
Qi Ou, Ping Tuo, Wenfei Li, Xiaoxu Wang, Yixiao Chen, Linfeng Zhang
J. Phys. Chem. C, 2023, 127 (37), 18755–18764.
DOI: 10.1021/acs.jpcc.3c04703
Truncated nonlocal kinetic energy density functionals for simple metals and silicon
Liang Sun, Yuanbo Li, Mohan Chen
Phys. Rev. B, 2023, 108 (7), 75158.
DOI: 10.1103/PhysRevB.108.075158
Interplay between magnetic structures and surface states in MnBi2Te4 from first-principles studies
Zujian Dai, Gan Jin, Lixin He
Phys. Rev. B, 2023, 108 (8), 85112.
DOI: 10.1103/PhysRevB.108.085112
Implementation of the meta-GGA exchange-correlation functional in numerical atomic orbital basis: With systematic testing on SCAN, rSCAN, and r2SCAN functionals
Renxi Liu, Daye Zheng, Xinyuan Liang, Xinguo Ren, Mohan Chen, Wenfei Li
J. Chem. Phys., 2023, 159 (7), 74109.
DOI: 10.1063/5.0160726
First-principles calculations of plasmon excitations in graphene, silicene, and germanene
Pengfei Li, Rong Shi, Peize Lin, Xinguo Ren
Phys. Rev. B, 2023, 107 (3), 35433.
DOI: 10.1103/PhysRevB.107.035433
2022
Disordered hyperuniform quasi-one-dimensional materials
Duyu Chen, Yu Liu, Yu Zheng, Houlong Zhuang, Mohan Chen, Yang Jiao
Phys. Rev. B, 2022, 106 (23), 235427.
DOI: 10.1103/PhysRevB.106.235427
DeePKS + ABACUS as a Bridge between Expensive Quantum Mechanical Models and Machine Learning Potentials
Wenfei Li, Qi Ou, Yixiao Chen, Yu Cao, Renxi Liu, Chunyi Zhang, Daye Zheng, Chun Cai, Xifan Wu, Han Wang, Mohan Chen, Linfeng Zhang
J. Phys. Chem. A, 2022, 126, 9154–9164.
DOI: 10.1021/acs.jpca.2c05000
First-principles calculations of the surface states of doped and alloyed topological materials via band unfolding method
Zujian Dai, Gan Jin, Lixin He
Comput. Mater. Sci., 2022, 213, 111656.
DOI: 10.1016/j.commatsci.2022.111656
Reproducibility of Hybrid Density Functional Calculations for Equation-of-State Properties and Band Gaps
Yuyang Ji, Peize Lin, Xinguo Ren, Lixin He
J. Phys. Chem., A, 2022, 126 (35), 5924–5931.
DOI: 10.1021/acs.jpca.2c05170
Plane-wave-based stochastic-deterministic density functional theory for extended systems
Qianrui Liu, Mohan Chen
Phys. Rev. B, 2022, 106 (12), 125132.
DOI: 10.1103/PhysRevB.106.125132
Density Functional Theory Plus Dynamical Mean Field Theory within the Framework of Linear Combination of Numerical Atomic Orbitals: Formulation and Benchmarks
Xin Qu, Peng Xu, Rusong Li, Gang Li, Lixin He, Xinguo Ren
J. Chem. Theory Comput., 2022, 18 (9), 5589–5606.
DOI: 10.1021/acs.jctc.2c00472
DFT+U within the framework of linear combination of numerical atomic orbitals
Xin Qu, Peng Xu, Hong Jiang, Lixin He, Xinguo Ren
J. Chem. Phys., 2022, 156 (23), 234104.
DOI: 10.1063/5.0090122
A caveat of the charge-extrapolation scheme for modeling electrochemical reactions on semiconductor surfaces: an issue induced by a discontinuous Fermi level change
Yu Liu, Xinlong Ding, Mohan Chen, Shenzhen Xu
Phys. Chem. Chem. Phys., 2022, 24 (25), 15511–15521.
DOI: 10.1039/D2CP00642A
Real-Time, Time-Dependent Density Functional Theory Study on Photoinduced Isomerizations of Azobenzene Under a Light Field
Fuxiang He, Xinguo Ren, Jun Jiang, Guozhen Zhang, Lixin He
J. Phys. Chem. Lett., 2022, 13 (2), 427–432.
DOI: 10.1021/acs.jpclett.1c03442
2021
Accurate stress calculations based on numerical atomic orbital bases: Implementation and benchmarks
Daye Zheng, Xinguo Ren, Lixin He
Comput. Phys. Commun., 2021, 267, 108043.
DOI: 10.1016/j.cpc.2021.108043
Peculiar diffusion behavior of AlCl4 intercalated in graphite from nanosecond-long molecular dynamics simulations*
Qianpeng Wang, Daye Zheng, Lixin He, Xinguo Ren
Chin. Phys, B, 2021, 30 (10), 107102.
DOI: 10.1088/1674-1056/ac0692
Calculation of Berry curvature using non-orthogonal atomic orbitals
Gan Jin, Daye Zheng, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2021, 33 (32), 325503.
DOI: 10.1088/1361-648X/ac05e5
Strategy for constructing compact numerical atomic orbital basis sets by incorporating the gradients of reference wavefunctions
Peize Lin, Xinguo Ren, Lixin He
Phys. Rev. B, 2021, 103 (23), 235131.
DOI: 10.1103/PhysRevB.103.235131
Copper-doped beryllium and beryllium oxide interface: A first- principles study
Yu Liu, Xiaohui Liu, Mohan Chen
J. Nucl. Mater., 2021, 545, 152733.
DOI: 10.1016/j.jnucmat.2020.152733
Beryllium and Magnesium Metal Clusters: New Globally Stable Structures and G0W0 Calculations
Sunila Bakhsh, Xiaohui Liu, Yanyong Wang, Lixin He, Xinguo Ren
J. Phys. Chem., A, 2021, 125 (7), 1424–1435.
DOI: 10.1021/acs.jpca.0c08960
Efficient Hybrid Density Functional Calculations for Large Periodic Systems Using Numerical Atomic Orbitals
Peize Lin, Xinguo Ren, Lixin He
J. Chem. Theory Comput., 2021, 17 (1), 222–239.
DOI: 10.1021/acs.jctc.0c00960
Retention and recycling of deuterium in liquid lithium-tin slab studied by first-principles molecular dynamics
Daye Zheng, Zhen-Xiong Shen, Mohan Chen, Xinguo Ren, Lixin He
J. Nucl. Mater., 2021, 543, 152542.
DOI: 10.1016/j.jnucmat.2020.152542
2020
Disordered hyperuniformity in two-dimensional amorphous silica
Yu Zheng, Lei Liu, Hanqing Nan, Zhen-Xiong Shen, Ge Zhang, Duyu Chen, Lixin He, Wenxiang Xu, Mohan Chen, Yang Jiao, Houlong Zhuang
Sci. Adv., 2020, 6 (16), eaba0826.
DOI: 10.1126/sciadv.aba0826
First-principles study of magnon-phonon interactions in gadolinium iron garnet
Lian-Wei Wang, Li-Shan Xie, Peng-Xiang Xu, Ke Xia
Phys. Rev. B, 2020, 101 (16), 165137.
DOI: 10.1103/PhysRevB.101.165137
Accuracy of Localized Resolution of the Identity in Periodic Hybrid Functional Calculations with Numerical Atomic Orbitals
Peize Lin, Xinguo Ren, Lixin He
J. Phys. Chem. Lett., 2020, 11 (8), 3082–3088.
DOI: 10.1021/acs.jpclett.0c00481
Optimizing the Void Size of Yolk-Shell Bi@Void@C Nanospheres for High- Power-Density Sodium-Ion Batteries
Hai Yang, Lin-Wei Chen, Fuxiang He, Jiaqing Zhang, Yuezhan Feng, Lukang Zhao, Bin Wang, Lixin He, Qiaobao Zhang, Yan Yu
Nano Lett., 2020, 20 (1), 758–767.
DOI: 10.1021/acs.nanolett.9b04829
2019
A DFT study of energetic and structural properties of a full turn of A-form DNA under relaxed and stretching conditions
Yue Liu, Xinguo Ren, Lixin He
J. Chem. Phys., 2019, 151 (21), 215102.
DOI: 10.1063/1.5129716
Cooperative Effect in a Graphite Intercalation Compound: Enhanced Mobility of AlCl4 in the Graphite Cathode of Aluminum-Ion Batteries
Qianpeng Wang, Daye Zheng, Lixin He, Xinguo Ren
Phys, Rev, Appl., 2019, 12 (4), 44060.
DOI: 10.1103/PhysRevApplied.12.044060
Structure evolution of chromium-doped boron clusters: toward the formation of endohedral boron cages
Xuecheng Shao, Xin Qu, Siyu Liu, Lihua Yang, Jinghai Yang, Xiaohui Liu, Xin Zhong, Shuai Sun, G. Vaitheeswaran, Jian Lv
Rsc Adv., 2019, 9 (5), 2870–2876.
DOI: 10.1039/C8RA09143A
2018
Diffusion coefficients of Mg isotopes in MgSiO3 and Mg2SiO4 melts calculated by first-principles molecular dynamics simulations
Xiaohui Liu, Yuhan Qi, Daye Zheng, Chen Zhou, Lixin He, Fang Huang
Geochim. Cosmochim. Acta, 2018, 223, 364–376.
DOI: 10.1016/j.gca.2017.12.007
2017
First-principles calculations and model analysis of plasmon excitations in graphene and graphene/hBN heterostructure
Pengfei Li, Xinguo Ren, Lixin He
Phys. Rev. B, 2017, 96 (16), 165417.
DOI: 10.1103/PhysRevB.96.165417
First-principles molecular dynamics study of deuterium diffusion in liquid tin
Xiaohui Liu, Daye Zheng, Xinguo Ren, Lixin He, Mohan Chen
J. Chem. Phys., 2017, 147 (6), 64505.
DOI: 10.1063/1.4997635
2016
Large-scale ab initio simulations based on systematically improvable atomic basis
Pengfei Li, Xiaohui Liu, Mohan Chen, Peize Lin, Xinguo Ren, Lin Lin, Chao Yang, Lixin He
Comput. Mater. Sci., 2016, 112, 503–517.
DOI: 10.1016/j.commatsci.2015.07.004
2013
Accelerating atomic orbital-based electronic structure calculation via pole expansion and selected inversion
Lin Lin, Mohan Chen, Chao Yang, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2013, 25 (29), 295501.
DOI: 10.1088/0953-8984/25/29/295501
2011
Electronic structure interpolation via atomic orbitals
Mohan Chen, G-C Guo, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2011, 23 (32), 325501.
DOI: 10.1088/0953-8984/23/32/325501
2010
Systematically improvable optimized atomic basis sets for ab initio calculations
Mohan Chen, G-C Guo, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2010, 22 (44), 445501.
DOI: 10.1088/0953-8984/22/44/445501
2009
Method to construct transferable minimal basis sets forab initiocalculations
Mohan Chen, Wei Fang, G.-Z. Sun, G.-C. Guo, Lixin He
Phys. Rev. B, 2009, 80 (16), 165121.
DOI: 10.1103/PhysRevB.80.165121