The following publications have used the DP-GEN software. Publications that only mentioned the DP-GEN will not be included below.
We encourage explicitly mentioning DP-GEN with proper citations in your publications, so we can more easily find and list these publications.
Last update date: June 24, 2025
2025
Temperature-driven structural phase transitions in SmNiO\$_3\$: insights from deep potential molecular dynamics simulations
Guoyong Shi, Fenglin Deng, Ri He, Dachuan Chen, Xuejiao Chen, Peiheng Jiang, Zhicheng Zhong
arXiv, 2025, 2503.06039.
DOI: 10.48550/arXiv.2503.06039
2024
Phase Transition in Silicon from Machine Learning Informed Metadynamics
Mangladeep Bhullar, Zihao Bai, Akinwumi Akinpelu, Yansun Yao
Chemphyschem: Eur. J. Chem. Phys. Phys. Chem., 2024, e202400090.
DOI: 10.1002/cphc.202400090
Non-equilibrium nature of fracture determines the crack paths
Pengjie Shi, Shizhe Feng, Zhiping Xu
Extrem. Mech. Lett., 2024, 68, 102151.
DOI: 10.1016/j.eml.2024.102151
Revisiting the phase diagram and piezoelectricity of lead zirconate titanate from first principles
Yubai Shi, Ri He, Bingwen Zhang, Zhicheng Zhong
Phys. Rev. B, 2024, 109 (17), 174104.
DOI: 10.1103/PhysRevB.109.174104
Exploring dielectric properties in atomistic models of amorphous boron nitride
Thomas Galvani, Ali K Hamze, Laura Caputo, Onurcan Kaya, Simon M-M Dubois, Luigi Colombo, Viet-Hung Nguyen, Yongwoo Shin, Hyeon-Jin Shin, Jean-Christophe Charlier, Stephan Roche
J. Phys. Mater., 2024, 7 (3), 35003.
DOI: 10.1088/2515-7639/ad4c06
Double-Layer Distribution of Hydronium and Hydroxide Ions in the Air-Water Interface
Pengchao Zhang, Muye Feng, Xuefei Xu
Acs Phys. Chem Au, 2024.
DOI: 10.1021/acsphyschemau.3c00076
Ultrafast switching dynamics of the ferroelectric order in stacking- engineered ferroelectrics
Ri He, Bingwen Zhang, Hua Wang, Lei Li, Ping Tang, Gerrit Bauer, Zhicheng Zhong
Acta Materialia, 2024, 262, 119416.
DOI: 10.1016/j.actamat.2023.119416
Unraveling pyrolysis mechanisms of lignin dimer model compounds: Neural network-based molecular dynamics simulation investigations
Zhe Shang, Hui Li
Fuel, 2024, 357, 129909.
DOI: 10.1016/j.fuel.2023.129909
2023
Active learning prediction and experimental confirmation of atomic structure and thermophysical properties for liquid Hf_{76}W_{24} refractory alloy
K. L. Liu, R. L. Xiao, Y. Ruan, B. Wei
Phys. Rev., E, 2023, 108 (5-2), 55310.
DOI: 10.1103/PhysRevE.108.055310
Universal interatomic potential for perovskite oxides
Jing Wu, Jiyuan Yang, Yuan-Jinsheng Liu, Duo Zhang, Yudi Yang, Yuzhi Zhang, Linfeng Zhang, Shi Liu
Phys. Rev. B, 2023, 108 (18), L180104.
DOI: 10.1103/PhysRevB.108.L180104
Efficient Molecular Dynamics Simulations of Deep Eutectic Solvents with First-Principles Accuracy Using Machine Learning Interatomic Potentials
Omid Shayestehpour, Stefan Zahn
J. Chem. Theory Comput., 2023, 19 (23), 8732–8742.
DOI: 10.1021/acs.jctc.3c00944
Ferroelectric Domain and Switching Dynamics in Curved In2Se3: First- Principles and Deep Learning Molecular Dynamics Simulations
Dongyu Bai, Yihan Nie, Jing Shang, Junxian Liu, Minghao Liu, Yang Yang, Haifei Zhan, Liangzhi Kou, Yuantong Gu
Nano Lett., 2023, 23 (23), 10922–10929.
DOI: 10.1021/acs.nanolett.3c03160
Data Efficient and Stability Indicated Sampling for Developing Reactive Machine Learning Potential to Achieve Ultralong Simulation in Lithium-Metal Batteries
Longkun Xu, Wei Shao, Haishun Jin, Qiang Wang
J. Phys. Chem. C, 2023, 127 (50), 24106–24117.
DOI: 10.1021/acs.jpcc.3c05522
Machine learning interatomic potential for molecular dynamics simulation of the ferroelectric KNbO3 perovskite
Hao-Cheng Thong, XiaoYang Wang, Jian Han, Linfeng Zhang, Bei Li, Ke Wang, Ben Xu
Phys. Rev. B, 2023, 107, 14101.
DOI: 10.1103/PhysRevB.107.014101
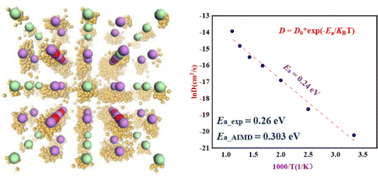
Li ion diffusion behavior of Li3OCl solid-state electrolytes with different defect structures: insights from the deep potential model
Zhou Zhang, Zhongyun Ma, Yong Pei
Phys. Chem. Chem. Phys., 2023, 25, 13297–13307.
DOI: 10.1039/d2cp06073f
A deep learning interatomic potential suitable for simulating radiation damage in bulk tungsten
Chang-Jie Ding, Ya-Wei Lei, Xiao-Yang Wang, Xiao-Lin Li, Xiang-Yan Li, Yan-Ge Zhang, Yi-Chun Xu, Chang-Song Liu, Xue-Bang Wu
Tungsten, 2023.
DOI: 10.1007/s42864-023-00230-4
Deep-learning potentials for proton transport in double-sided graphanol
Siddarth K. Achar, Leonardo Bernasconi, Juan J. Alvarez, J. Karl Johnson
Journal of Materials Research, 2023.
DOI: 10.1557/s43578-023-01141-3
Speciation of La3+-Cl- Complexes in Hydrothermal Fluids from Deep Potential Molecular Dynamics
Wei Zhang, Li Zhou, Tinggui Yan, Mohan Chen
J. Phys. Chem. B, 2023, 127, 8926–8937.
DOI: 10.1021/acs.jpcb.3c05428
Neural Network Water Model Based on the MB-Pol Many-Body Potential
Maria Carolina Muniz, Roberto Car, Athanassios Z Panagiotopoulos
J. Phys. Chem. B, 2023, 127, 9165–9171.
DOI: 10.1021/acs.jpcb.3c04629
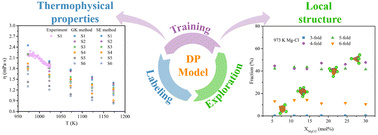
Insights into the local structure evolution and thermophysical properties of NaCl-KCl-MgCl2\texte ndashLaCl3 melt driven by machine learning
Jia Zhao, Taixi Feng, Guimin Lu, Jianguo Yu
J. Mater. Chem. A, 2023, 11, 23999–24012.
DOI: 10.1039/d3ta03434h
Constrained Hybrid Monte Carlo Sampling Made Simple for Chemical Reaction Simulations
Bin Jin, Taiping Hu, Kuang Yu, Shenzhen Xu
J. Chem. Theory Comput., 2023, 19, 7343–7357.
DOI: 10.1021/acs.jctc.3c00571
Machine learning assisted investigation of the barocaloric performance in ammonium iodide
Xiong Xu, Fangbiao Li, Chang Niu, Min Li, Hui Wang
2023, 122.
DOI: 10.1063/5.0131696
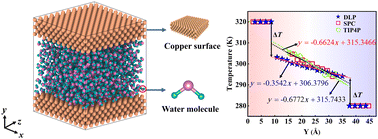
Thermal transport across copper-water interfaces according to deep potential molecular dynamics
Zhiqiang Li, Xiaoyu Tan, Zhiwei Fu, Linhua Liu, Jia-Yue Yang
Phys. Chem. Chem. Phys., 2023, 25, 6746–6756.
DOI: 10.1039/d2cp05530a
Structure and solidification of the (Fe0.75B0.15Si0.1)100-xTax (x=0-2) melts: Experiment and machine learning
I.V. Sterkhova, L.V. Kamaeva, V.I. Lad'yanov, N.M. Chtchelkatchev
Journal of Physics and Chemistry of Solids, 2023, 174, 111143.
DOI: 10.1016/j.jpcs.2022.111143
Unravelling the dissolution dynamics of silicate minerals by deep learning molecular dynamics simulation: A case of dicalcium silicate
Yunjian Li, Hui Pan, Zongjin Li
Cement and Concrete Research, 2023, 165, 107092.
DOI: 10.1016/j.cemconres.2023.107092
Profiling the off-center atomic displacements in CuCl at finite temperatures with a deep-learning potential
Zhi-Hao Wang, Xuan-Yan Chen, Zhen Zhang, Xie Zhang, Su- Huai Wei
Phys. Rev. Materials, 2023, 7, 34601.
DOI: 10.1103/PhysRevMaterials.7.034601
Liquid-Crystal Structure Inheritance in Machine Learning Potentials for Network-Forming Systems
I. A. Balyakin, R. E. Ryltsev, N. M. Chtchelkatchev
Jetp Lett., 2023, 117, 370–376.
DOI: 10.1134/S0021364023600234
Atomic structure, stability, and dissociation of dislocations in cadmium telluride
Jun Li, Kun Luo, Qi An
International Journal of Plasticity, 2023, 163, 103552.
DOI: 10.1016/j.ijplas.2023.103552
The highest melting point material: Searched by Bayesian global optimization with deep potential molecular dynamics
Yinan Wang, Bo Wen, Xingjian Jiao, Ya Li, Lei Chen, Yujin Wang, Fu-Zhi Dai
2023, 12, 803–814.
DOI: 10.26599/JAC.2023.9220721
Structural and Composition Evolution of Palladium Catalyst for CO Oxidation under Steady-State Reaction Conditions
Jiawei Wu, Dingming Chen, Jianfu Chen, Haifeng Wang
J. Phys. Chem. C, 2023, 127, 6262–6270.
DOI: 10.1021/acs.jpcc.2c07877
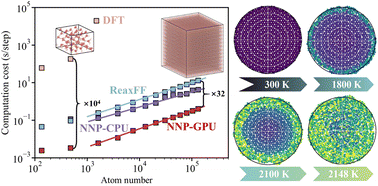
Monitoring the melting behavior of boron nanoparticles using a neural network potential
Xiaoya Chang, Qingzhao Chu, Dongping Chen
Phys. Chem. Chem. Phys., 2023, 25, 12841–12853.
DOI: 10.1039/d3cp00571b
Unraveling the Dynamic Correlations between Transition Metal Migration and the Oxygen Dimer Formation in the Highly Delithiated LixCoO2 Cathode
Taiping Hu, Fu-Zhi Dai, Guobing Zhou, Xiaoxu Wang, Shenzhen Xu
J. Phys. Chem. Lett., 2023, 14, 3677–3684.
DOI: 10.1021/acs.jpclett.3c00506
Hydrogen distribution between the Earth's inner and outer core
Liang Yuan, Gerd Steinle-Neumann
Earth and Planetary Science Letters, 2023, 609, 118084.
DOI: 10.1016/j.epsl.2023.118084
Lattice Thermal Conductivity of Monolayer InSe Calculated by Machine Learning Potential
Jinsen Han, Qiyu Zeng, Ke Chen, Xiaoxiang Yu, Jiayu Dai
Nanomaterials (Basel)., 2023, 13, 1576.
DOI: 10.3390/nano13091576
Modeling the high-pressure solid and liquid phases of tin from deep potentials with ab initio accuracy
Tao Chen, Fengbo Yuan, Jianchuan Liu, Huayun Geng, Linfeng Zhang, Han Wang, Mohan Chen
Phys. Rev. Materials, 2023, 7, 53603.
DOI: 10.1103/PhysRevMaterials.7.053603
A Deep Potential model for liquid-vapor equilibrium and cavitation rates of water
Ignacio Sanchez-Burgos, Maria Carolina Muniz, Jorge R Espinosa, Athanassios Z Panagiotopoulos
J. Chem. Phys., 2023, 158.
DOI: 10.1063/5.0144500
First-Principles-Based Machine Learning Models for Phase Behavior and Transport Properties of CO2
Reha Mathur, Maria Carolina Muniz, Shuwen Yue, Roberto Car, Athanassios Z Panagiotopoulos
J. Phys. Chem. B, 2023, 127, 4562–4569.
DOI: 10.1021/acs.jpcb.3c00610
An accurate interatomic potential for the TiAlNb ternary alloy developed by deep neural network learning method
Jiajun Lu, Jinkai Wang, Kaiwei Wan, Ying Chen, Hao Wang, Xinghua Shi
J. Chem. Phys., 2023, 158.
DOI: 10.1063/5.0147720
In Silico Demonstration of Fast Anhydrous Proton Conduction on Graphanol
Siddarth K Achar, Leonardo Bernasconi, Ruby I DeMaio, Katlyn R Howard, J Karl Johnson
ACS Appl. Mater. Interfaces, 2023, 15, 25873–25883.
DOI: 10.1021/acsami.3c04022
Noble gas (He, Ne, and Ar) solubilities in high-pressure silicate melts calculated based on deep-potential modeling
Kai Wang, Xiancai Lu, Xiandong Liu, Kun Yin
Geochimica et Cosmochimica Acta, 2023, 350, 57–68.
DOI: 10.1016/j.gca.2023.03.032
Deciphering the Anomalous Acidic Tendency of Terminal Water at Rutile(110)-Water Interfaces
Yong-Bin Zhuang, Jun Cheng
J. Phys. Chem. C, 2023, 127, 10532–10540.
DOI: 10.1021/acs.jpcc.3c01870
Investigating the Hydroxyl Reorientation in Hydroxyapatite Using Machine Learning Potentials
Jing Wang, Xin Wang, Hua Zhu, Dingguo Xu
J. Phys. Chem. C, 2023, 127, 11369–11377.
DOI: 10.1021/acs.jpcc.3c02426
Molecular insight into the GaP(110)-water interface using machine learning accelerated molecular dynamics
Xue-Ting Fan, Xiao-Jian Wen, Yong-Bin Zhuang, Jun Cheng
Journal of Energy Chemistry, 2023, 82, 239–247.
DOI: 10.1016/j.jechem.2023.03.013
Revealing Carbon Vacancy Distribution on $\alpha$-MoC1-x Surfaces by Machine-Learning Force-Field-Aided Cluster Expansion Approach
Jun-Zhong Xie, Hong Jiang
J. Phys. Chem. C, 2023, 127, 13228–13237.
DOI: 10.1021/acs.jpcc.3c01941
Fluorine spillover for ceria- vs silica-supported palladium nanoparticles: A MD study using machine learning potentials
Da-Jiang Liu, James W Evans
J. Chem. Phys., 2023, 159.
DOI: 10.1063/5.0147132
Local structure, thermodynamics, and melting of boron phosphide at high pressures by deep learning-driven ab~initio simulations
N M Chtchelkatchev, R E Ryltsev, M V Magnitskaya, S M Gorbunov, K A Cherednichenko, V L Solozhenko, V V Brazhkin
J. Chem. Phys., 2023, 159.
DOI: 10.1063/5.0165948
Deep Potential Molecular Dynamics Study of Chapman-Jouguet Detonation Events of Energetic Materials
Jidong Zhang, Wei Guo, Yugui Yao
J. Phys. Chem. Lett., 2023, 14, 7141–7148.
DOI: 10.1021/acs.jpclett.3c01392
Deep neural network potential for simulating hydrogen blistering in tungsten
Xiao-Yang Wang, Yi-Nan Wang, Ke Xu, Fu-Zhi Dai, Hai-Feng Liu, Guang-Hong Lu, Han Wang
Phys. Rev. Materials, 2023, 7, 93601.
DOI: 10.1103/PhysRevMaterials.7.093601
Unraveling the Atomic-scale Mechanism of Phase Transformations and Structural Evolutions during (de)Lithiation in Si Anodes
Fangjia Fu, Xiaoxu Wang, Linfeng Zhang, Yifang Yang, Jianhui Chen, Bo Xu, Chuying Ouyang, Shenzhen Xu, Fu-Zhi Dai, Weinan E
Adv Funct Materials, 2023, 33.
DOI: 10.1002/adfm.202303936
Collective motion in hcp-Fe at Earth\textquoterights inner core conditions
Youjun Zhang, Yong Wang, Yuqian Huang, Junjie Wang, Zhixin Liang, Long Hao, Zhipeng Gao, Jun Li, Qiang Wu, Hong Zhang, Yun Liu, Jian Sun, Jung-Fu Lin
Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2309952120.
DOI: 10.1073/pnas.2309952120
Machine learning potential for Ab Initio phase transitions of zirconia
Yuanpeng Deng, Chong Wang, Xiang Xu, Hui Li
Theoretical and Applied Mechanics Letters, 2023, 13, 100481.
DOI: 10.1016/j.taml.2023.100481
Modelling electrified microporous carbon/electrolyte electrochemical interface and unraveling charge storage mechanism by machine learning accelerated molecular dynamics
Yifeng Zhang, Hui Huang, Jie Tian, Chengwei Li, Yuchen Jiang, Zeng Fan, Lujun Pan
Energy Storage Materials, 2023, 63, 103069.
DOI: 10.1016/j.ensm.2023.103069
Data-driven prediction of complex crystal structures of dense lithium
Xiaoyang Wang, Zhenyu Wang, Pengyue Gao, Chengqian Zhang, Jian Lv, Han Wang, Haifeng Liu, Yanchao Wang, Yanming Ma
Nat. Commun., 2023, 14, 2924.
DOI: 10.1038/s41467-023-38650-y

Realizing long-cycling all-solid-state Li-In||TiS2 batteries using Li6+xMxAs1-xS5I (M=Si, Sn) sulfide solid electrolytes
Pushun Lu, Yu Xia, Guochen Sun, Dengxu Wu, Siyuan Wu, Wenlin Yan, Xiang Zhu, Jiaze Lu, Quanhai Niu, Shaochen Shi, Zhengju Sha, Liquan Chen, Hong Li, Fan Wu
Nat. Commun., 2023, 14, 4077.
DOI: 10.1038/s41467-023-39686-w
Dislocation-mediated migration of the $\alpha$/$\beta$ interfaces in titanium
Jin-Yu Zhang, Zhi-Peng Sun, Dong Qiu, Fu-Zhi Dai, Yang- Sheng Zhang, Dongsheng Xu, Wen-Zheng Zhang
Acta Materialia, 2023, 261, 119364.
DOI: 10.1016/j.actamat.2023.119364
Interfacial heat and mass transfer at silica/binary molten salt interface from deep potential molecular dynamics
Fei Liang, Jing Ding, Xiaolan Wei, Gechuanqi Pan, Shule Liu
International Journal of Heat and Mass Transfer, 2023, 217, 124705.
DOI: 10.1016/j.ijheatmasstransfer.2023.124705
Revisiting the structure, interaction, and dynamical property of ionic liquid from the deep learning force field
Yulong Ling, Kun Li, Mi Wang, Junfeng Lu, Chenlu Wang, Yanlei Wang, Hongyan He
Journal of Power Sources, 2023, 555, 232350.
DOI: 10.1016/j.jpowsour.2022.232350
Solvation structures of calcium and magnesium ions in water with the presence of hydroxide: a study by deep potential molecular dynamics
Jianchuan Liu, Renxi Liu, Yu Cao, Mohan Chen
Phys. Chem. Chem. Phys., 2023.
DOI: 10.1039/d2cp04105g
Accurate interatomic potential for the nucleation in liquid Ti-Al binary alloy developed by deep neural network learning method
B. Zhai, H.P. Wang
Computational Materials Science, 2023, 216, 111843.
DOI: 10.1016/j.commatsci.2022.111843
2022
Convergence acceleration in machine learning potentials for atomistic simulations
Dylan Bayerl, Christopher M. Andolina, Shyam Dwaraknath, Wissam A. Saidi
Digital Discovery, 2022, 1, 61–69.
DOI: 10.1039/d1dd00005e
Towards fully ab initio simulation of atmospheric aerosol nucleation
Shuai Jiang, Yi-Rong Liu, Teng Huang, Ya-Juan Feng, Chun- Yu Wang, Zhong-Quan Wang, Bin-Jing Ge, Quan-Sheng Liu, Wei-Ran Guang, Wei Huang
Nat. Commun., 2022, 13, 6067.
DOI: 10.1038/s41467-022-33783-y
Modeling Chemical Reactions in Alkali Carbonate-Hydroxide Electrolytes with Deep Learning Potentials
Anirban Mondal, Dina Kussainova, Shuwen Yue, Athanassios Z Panagiotopoulos
J. Chem. Theory Comput., 2022.
DOI: 10.1021/acs.jctc.2c00816
Lattice Thermal Conductivity of MgSiO3 Perovskite and Post- Perovskite under Lower Mantle Conditions Calculated by Deep Potential Molecular Dynamics
Fenghu Yang, Qiyu Zeng, Bo Chen, Dongdong Kang, Shen Zhang, Jianhua Wu, Xiaoxiang Yu, Jiayu Dai
Chinese Phys. Lett., 2022, 39, 116301.
DOI: 10.1088/0256-307X/39/11/116301
Resolving the odd-even oscillation of water dissociation at rutile TiO2(110)-water interface by machine learning accelerated molecular dynamics
Yong-Bin Zhuang, Rui-Hao Bi, Jun Cheng
J. Chem. Phys., 2022, 157, 164701.
DOI: 10.1063/5.0126333
Origin of negative thermal expansion and pressure-induced amorphization in zirconium tungstate from a machine-learning potential
Ri He, Hongyu Wu, Yi Lu, Zhicheng Zhong
Phys. Rev. B, 2022, 106, 174101.
DOI: 10.1103/PhysRevB.106.174101
Classical and machine learning interatomic potentials for BCC vanadium
Rui Wang, Xiaoxiao Ma, Linfeng Zhang, Han Wang, David J. Srolovitz, Tongqi Wen, Zhaoxuan Wu
Phys. Rev. Materials, 2022, 6, 113603.
DOI: 10.1103/PhysRevMaterials.6.113603
Metal Affinity of Support Dictates Sintering of Gold Catalysts
Jin-Cheng Liu, Langli Luo, Hai Xiao, Junfa Zhu, Yang He, Jun Li
J. Am. Chem. Soc., 2022, 144, 20601–20609.
DOI: 10.1021/jacs.2c06785
Multireference Generalization of the Weighted Thermodynamic Perturbation Method
Timothy J Giese, Jinzhe Zeng, Darrin M York
J. Phys. Chem. A, 2022, 126, 8519–8533.
DOI: 10.1021/acs.jpca.2c06201
Thermal Conductivity of Hydrous Wadsleyite Determined by Non-Equilibrium Molecular Dynamics Based on Machine Learning
Dong Wang, Zhongqing Wu, Xin Deng
Geophysical Research Letters, 2022, 49.
DOI: 10.1029/2022GL100337
Deep potential for a face-centered cubic Cu system at finite temperatures
Yunzhen Du, Zhaocang Meng, Qiang Yan, Canglong Wang, Yuan Tian, Wenshan Duan, Sheng Zhang, Ping Lin
Phys. Chem. Chem. Phys., 2022, 24, 18361–18369.
DOI: 10.1039/D2CP02758E
Structural and electrocatalytic properties of copper clusters: A study via deep learning and first principles
Xiaoning Wang, Haidi Wang, Qiquan Luo, Jinlong Yang
J. Chem. Phys., 2022, 157, 74304.
DOI: 10.1063/5.0100505
High accuracy neural network interatomic potential for NiTi shape memory alloy
Hao Tang, Yin Zhang, Qing-Jie Li, Haowei Xu, Yuchi Wang, Yunzhi Wang, Ju Li
Acta Materialia, 2022, 238, 118217.
DOI: 10.1016/j.actamat.2022.118217
A tungsten deep neural-network potential for simulating mechanical property degradation under fusion service environment
Xiaoyang Wang, Yinan Wang, Linfeng Zhang, Fuzhi Dai, Han Wang
Nucl. Fusion, 2022, 62, 126013.
DOI: 10.1088/1741-4326/ac888b
Molecular dynamics simulations of LiCl ion pairs in high temperature aqueous solutions by deep learning potential
Wei Zhang, Li Zhou, Bin Yang, Tinggui Yan
Journal of Molecular Liquids, 2022, 367, 120500.
DOI: 10.1016/j.molliq.2022.120500
Combined QM/MM, Machine Learning Path Integral Approach to Compute Free Energy Profiles and Kinetic Isotope Effects in RNA Cleavage Reactions
Timothy J Giese, Jinzhe Zeng, Şölen Ekesan, Darrin M York
J. Chem. Theory Comput., 2022, 18, 4304–4317.
DOI: 10.1021/acs.jctc.2c00151
Machine Learning Accelerates Molten Salt Simulations: Thermal Conductivity of MgCl 2 -NaCl Eutectic
Wenshuo Liang, Guimin Lu, Jianguo Yu
Advcd Theory and Sims, 2022, 2200206.
DOI: 10.1002/adts.202200206
Machine Learning Force Field Aided Cluster Expansion Approach to Configurationally Disordered Materials: Critical Assessment of Training Set Selection and Size Convergence
Jun-Zhong Xie, Xu-Yuan Zhou, Dong Luan, Hong Jiang
J. Chem. Theory Comput., 2022, 18, 3795–3804.
DOI: 10.1021/acs.jctc.2c00017
Combined Deep Learning and Classical Potential Approach for Modeling Diffusion in UiO-66
Siddarth K Achar, Jacob J Wardzala, Leonardo Bernasconi, Linfeng Zhang, J Karl Johnson
J. Chem. Theory Comput., 2022, 18, 3593–3606.
DOI: 10.1021/acs.jctc.2c00010
Towards large-scale and spatiotemporally resolved diagnosis of electronic density of states by deep learning
Qiyu Zeng, Bo Chen, Xiaoxiang Yu, Shen Zhang, Dongdong Kang, Han Wang, Jiayu Dai
Phys. Rev. B, 2022, 105, 174109.
DOI: 10.1103/PhysRevB.105.174109
Exploring Complex Reaction Networks Using Neural Network-Based Molecular Dynamics Simulation
Qingzhao Chu, Kai H Luo, Dongping Chen
J. Phys. Chem. Lett., 2022, 13, 4052–4057.
DOI: 10.1021/acs.jpclett.2c00647
Acids at the Edge: Why Nitric and Formic Acid Dissociations at Air-Water Interfaces Depend on Depth and on Interface Specific Area
Miguel de la Puente, Rolf David, Axel Gomez, Damien Laage
J. Am. Chem. Soc., 2022, 144, 10524–10529.
DOI: 10.1021/jacs.2c03099
Dissolving salt is not equivalent to applying a pressure on water
Chunyi Zhang, Shuwen Yue, Athanassios Z Panagiotopoulos, Michael L Klein, Xifan Wu
Nat. Commun., 2022, 13, 822.
DOI: 10.1038/s41467-022-28538-8
Temperature- and vacancy-concentration-dependence of heat transport in Li3ClO from multi-method numerical simulations
Paolo Pegolo, Stefano Baroni, Federico Grasselli
npj Comput Mater, 2022, 8, 24.
DOI: 10.1038/s41524-021-00693-4
The chemical origin of temperature-dependent lithium-ion concerted diffusion in sulfide solid electrolyte Li10GeP2S12
Zhong-Heng Fu, Xiang Chen, Nan Yao, Xin Shen, Xia-Xia Ma, Shuai Feng, Shuhao Wang, Rui Zhang, Linfeng Zhang, Qiang Zhang
Journal of Energy Chemistry, 2022, 70, 59–66.
DOI: 10.1016/j.jechem.2022.01.018

Efficient and accurate atomistic modeling of dopant migration using deep neural network
Xi Ding, Ming Tao, Junhua Li, Mingyuan Li, Mengchao Shi, Jiashu Chen, Zhen Tang, Francis Benistant, Jie Liu
Materials Science in Semiconductor Processing, 2022, 143, 106513.
DOI: 10.1016/j.mssp.2022.106513
Exploring the Effects of Ionic Defects on the Stability of CsPbI 3 with a Deep Learning Potential
Weijie Yang, Jiajia Li, Xuelu Chen, Yajun Feng, Chongchong Wu, Ian D Gates, Zhengyang Gao, Xunlei Ding, Jianxi Yao, Hao Li
Chemphyschem, 2022, 23, e202100841.
DOI: 10.1002/cphc.202100841

Self-Healing Mechanism of Lithium in Lithium Metal
Junyu Jiao, Genming Lai, Liang Zhao, Jiaze Lu, Qidong Li, Xianqi Xu, Yao Jiang, Yan-Bing He, Chuying Ouyang, Feng Pan, Hong Li, Jiaxin Zheng
Adv. Sci. (Weinh)., 2022, 9, e2105574.
DOI: 10.1002/advs.202105574
A deep potential model with long-range electrostatic interactions
Linfeng Zhang, Han Wang, Maria Carolina Muniz, Athanassios Z Panagiotopoulos, Roberto Car, Weinan E
J. Chem. Phys., 2022, 156, 124107.
DOI: 10.1063/5.0083669
A generalizable machine learning potential of Ag-Au nanoalloys and its application to surface reconstruction, segregation and diffusion
YiNan Wang, LinFeng Zhang, Ben Xu, XiaoYang Wang, Han Wang
Modelling Simul. Mater. Sci. Eng., 2022, 30, 25003.
DOI: 10.1088/1361-651X/ac4002

Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water
Manyi Yang, Luigi Bonati, Daniela Polino, Michele Parrinello
Catalysis Today, 2022, 387, 143–149.
DOI: 10.1016/j.cattod.2021.03.018
Molecular dynamics simulation of molten strontium chloride based on deep potential
Di Guo, Jia Zhao, Wenshuo Liang, Guimin Lu
Journal of Molecular Liquids, 2022, 348, 118380.
DOI: 10.1016/j.molliq.2021.118380
Structural phase transitions in $\mathrmSrTi\mathrmO_3$ from deep potential molecular dynamics
Ri He, Hongyu Wu, Linfeng Zhang, Xiaoxu Wang, Fangjia Fu, Shi Liu, Zhicheng Zhong
Phys. Rev. B, 2022, 105, 064104.
DOI: 10.1103/PhysRevB.105.064104
A deep learning potential applied in tobermorite phases and extended to calcium silicate hydrates
Yang Zhou, Haojie Zheng, Weihuan Li, Tao Ma, Changwen Miao
Cement and Concrete Research, 2022, 152, 106685.
DOI: 10.1016/j.cemconres.2021.106685
2021

Insights from Computational Studies on the Anisotropic Volume Change of LixNiO2 at High States of Charge (x < 0.25)
Juan C. Garcia, Joshua Gabriel, Noah H. Paulson, John Low, Marius Stan, Hakim Iddir
J. Phys. Chem. C, 2021, 125 (49), 27130-27139.
DOI: 10.1021/acs.jpcc.1c08022
Specialising neural network potentials for accurate properties and application to the mechanical response of titanium
Tongqi Wen, Rui Wang, Lingyu Zhu, Linfeng Zhang, Han Wang, David J. Srolovitz, Zhaoxuan Wu
npj Comput Mater, 2021, 7, 206.
DOI: 10.1038/s41524-021-00661-y

Artificial intelligence model for efficient simulation of monatomic phase change material antimony
Mengchao Shi, Junhua Li, Ming Tao, Xin Zhang, Jie Liu
Materials Science in Semiconductor Processing, 2021, 136, 106146.
DOI: 10.1016/j.mssp.2021.106146

Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/Molecular Mechanical Simulations of Chemical Reactions in Solution
Jinzhe Zeng, Timothy J Giese, Şölen Ekesan, Darrin M York
J. Chem. Theory Comput., 2021, 17, 6993–7009.
DOI: 10.1021/acs.jctc.1c00201
Accurate force field of two-dimensional ferroelectrics from deep learning
Jing Wu, Liyi Bai, Jiawei Huang, Liyang Ma, Jian Liu, Shi Liu
Phys. Rev. B, 2021, 104, 174107.
DOI: 10.1103/PhysRevB.104.174107
Liquid-Liquid Critical Point in Phosphorus
Manyi Yang, Tarak Karmakar, Michele Parrinello
Phys. Rev. Lett., 2021, 127, 80603.
DOI: 10.1103/PhysRevLett.127.080603
Anomalous Behavior of Viscosity and Electrical Conductivity of MgSiO 3 Melt at Mantle Conditions
Haiyang Luo, Bijaya B. Karki, Dipta B. Ghosh, Huiming Bao
Geophys Res Lett, 2021, 48.
DOI: 10.1029/2021GL093573
Phase Diagram of a Deep Potential Water Model
Linfeng Zhang, Han Wang, Roberto Car, Weinan E
Phys. Rev. Lett., 2021, 126, 236001.
DOI: 10.1103/PhysRevLett.126.236001
The thermoelectric performance of new structure SnSe studied by quotient graph and deep learning potential
D. Guo, C. Li, K. Li, B. Shao, D. Chen, Y. Ma, J. Sun, X. Cao, W. Zeng, X. Chang
Materials Today Energy, 2021, 20, 100665.
DOI: 10.1016/j.mtener.2021.100665
Phase Equilibrium of Water with Hexagonal and Cubic Ice Using the SCAN Functional
Pablo M Piaggi, Athanassios Z Panagiotopoulos, Pablo G Debenedetti, Roberto Car
J. Chem. Theory Comput., 2021, 17, 3065–3077.
DOI: 10.1021/acs.jctc.1c00041
Accurate Deep Potential model for the Al-Cu-Mg alloy in the full concentration space*
Wanrun Jiang, Yuzhi Zhang, Linfeng Zhang, Han Wang
Chinese Phys. B, 2021, 30, 50706.
DOI: 10.1088/1674-1056/abf134

Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors
Jianxing Huang, Linfeng Zhang, Han Wang, Jinbao Zhao, Jun Cheng, Weinan E
J. Chem. Phys., 2021, 154, 94703.
DOI: 10.1063/5.0041849
Anharmonic Raman spectra simulation of crystals from deep neural networks
Honghui Shang, Haidi Wang
AIP Advances, 2021, 11, 35105.
DOI: 10.1063/5.0040190
Deep learning of accurate force field of ferroelectric HfO2
Jing Wu, Yuzhi Zhang, Linfeng Zhang, Shi Liu
Phys. Rev. B, 2021, 103, 24108.
DOI: 10.1103/PhysRevB.103.024108

Theoretical study of Na+ transport in the solid-state electrolyte Na3OBr based on deep potential molecular dynamics
Han-Xiao Li, Xu-Yuan Zhou, Yue-Chao Wang, Hong Jiang
Inorg. Chem. Front., 2021, 8, 425–432.
DOI: 10.1039/D0QI00921K

Exploring the Chemical Space of Linear Alkane Pyrolysis via Deep Potential GENerator
Jinzhe Zeng, Linfeng Zhang, Han Wang, Tong Zhu
Energy & Fuels, 2021, 35 (1), 762–769.
DOI: 10.1021/acs.energyfuels.0c03211
Diffusional fractionation of helium isotopes in silicate melts
H. Luo, B.B. Karki, D.B. Ghosh, H. Bao
Geochem. Persp. Let., 2021, 19–22.
DOI: 10.7185/geochemlet.2128
Deep-learning potential method to simulate shear viscosity of liquid aluminum at high temperature and high pressure by molecular dynamics
Yuqing Cheng, Han Wang, Shuaichuang Wang, Xingyu Gao, Qiong Li, Jun Fang, Hongzhou Song, Weidong Chu, Gongmu Zhang, Haifeng Song, Haifeng Liu
AIP Advances, 2021, 11, 15043.
DOI: 10.1063/5.0036298
2020
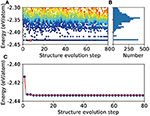
Crystal Structure Prediction of Binary Alloys via Deep Potential
Haidi Wang, Yuzhi Zhang, Linfeng Zhang, Han Wang
Front. Chem., 2020, 8, 589795.
DOI: 10.3389/fchem.2020.589795

Signatures of a liquid-liquid transition in an ab initio deep neural network model for water
Thomas E Gartner 3rd, Linfeng Zhang, Pablo M Piaggi, Roberto Car, Athanassios Z Panagiotopoulos, Pablo G Debenedetti
Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26040–26046.
DOI: 10.1073/pnas.2015440117

A Deep-Learning Potential for Crystalline and Amorphous Li-Si Alloys
Nan Xu, Yao Shi, Yi He, Qing Shao
J. Phys. Chem. C, 2020, 124, 16278–16288.
DOI: 10.1021/acs.jpcc.0c03333
Deep neural network for the dielectric response of insulators
Linfeng Zhang, Mohan Chen, Xifan Wu, Han Wang, Weinan E, Roberto Car
Phys. Rev. B, 2020, 102, 41121.
DOI: 10.1103/PhysRevB.102.041121
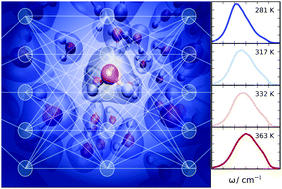
Raman spectrum and polarizability of liquid water from deep neural networks
Grace M Sommers, Marcos F Calegari Andrade, Linfeng Zhang, Han Wang, Roberto Car
Phys. Chem. Chem. Phys., 2020, 22, 10592–10602.
DOI: 10.1039/D0CP01893G

DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models
Yuzhi Zhang, Haidi Wang, Weijie Chen, Jinzhe Zeng, Linfeng Zhang, Han Wang, Weinan E
Computer Physics Communications, 2020, 253, 107206.
DOI: 10.1016/j.cpc.2020.107206