Uni-Mol: Partnering with the DeepModeling Community to Build a Foundational Model for Molecular Design
Pre-trained models are sweeping through the AI field by extracting representative information from large-scale unlabeled data and then performing supervised learning on small-scale labeled downstream tasks, becoming the de facto solution in many application scenarios. In drug design, there is still no consensus on the "best way to represent molecules." In the field of materials chemistry, predicting molecular properties is equally important. Mainstream molecular pre-training models typically start from one-dimensional sequences or two-dimensional graph structures, but molecular structures are inherently represented in three-dimensional space. Therefore, directly constructing pre-trained models from three-dimensional information to achieve better molecular representations has become an important and meaningful problem. To further promote research on molecular representation and pre-trained models, Uni-Mol will join the DeepModeling community to work with community developers to advance the development of a three-dimensional molecular representation pre-training framework.
Background of Pre-trained Models and Molecular Representation Learning
Pre-trained models achieve efficient model training and performance improvement by extracting representative information from large-scale unlabeled data and then performing supervised learning on small-scale labeled downstream tasks. Especially in fields like natural language processing and computer vision, pre-trained models have shown significant advantages.
In the field of molecular representation learning, researchers have also begun to use pre-trained models to enhance molecular representation capabilities. Traditional molecular pre-trained models mainly start from one-dimensional sequences (such as SMILES strings) or two-dimensional graph structures (such as molecular topology graphs). However, molecular structures are inherently represented in three-dimensional space, and ignoring this high-dimensional information limits the expressive power and application range of existing methods. Especially in fields like drug design, although these models perform well in limited supervised data learning, their performance is not ideal when dealing with tasks related to three-dimensional geometry.
Therefore, constructing pre-trained models directly from three-dimensional information to obtain more accurate and comprehensive molecular representations has become an important and meaningful research problem. This approach can better capture the three-dimensional characteristics of molecular structures and play a greater role in downstream tasks such as geometric prediction and molecular generation. With advances in computational resources and algorithmic technologies, three-dimensional molecular pre-trained models are gradually becoming a new hotspot in molecular representation learning research, driving the field forward.
This research background, which integrates pre-trained models and molecular representation learning, demonstrates the broad application potential of AI technology in scientific research, providing new ideas and tools for future molecular design and materials science research. Accordingly, we have developed Uni-Mol, a general three-dimensional molecular representation learning framework that greatly expands the expressive power and application range of the method.
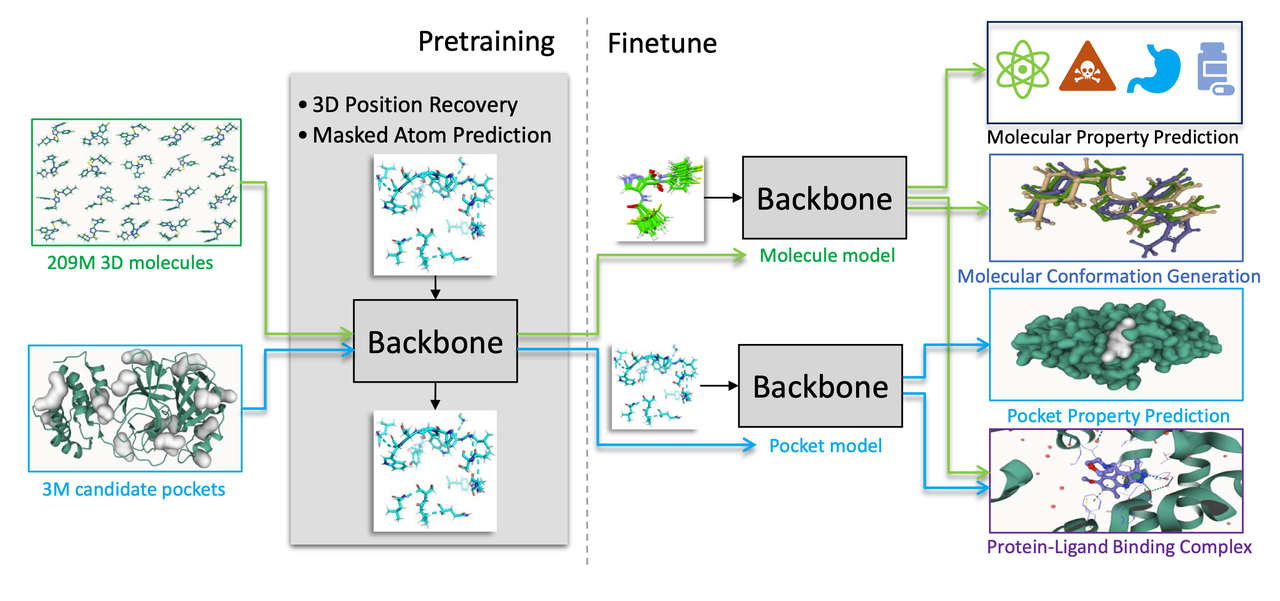
Introduction to Uni-Mol
In May 2022, the open-source general 3D molecular representation learning framework based on molecular three-dimensional structure, Uni-Mol, was officially launched, and the paper was accepted by the top machine learning conference ICLR 2023. Unlike previous molecular representation frameworks based on one-dimensional sequences or two-dimensional graph structures, Uni-Mol directly uses the three-dimensional structure of molecules as model input. Uni-Mol excels in performance and model generalization ability, surpassing existing solutions in tasks such as small molecule property prediction, protein target prediction, protein-ligand complex conformation prediction, quantum chemical property prediction, MOF material adsorption performance prediction, and OLED luminescent material performance prediction.
The Uni-Mol framework mainly includes two models: a molecular pre-training model trained using the three-dimensional conformations of 209 million molecules, and a pocket pre-training model trained using 3 million candidate protein data. These two models can be used independently for different tasks or combined for protein-ligand binding tasks. Uni-Mol outperforms current state-of-the-art techniques in 14 out of 15 molecular property prediction tasks. Additionally, Uni-Mol delivers excellent prediction results in three-dimensional space tasks such as protein-ligand binding mode prediction and molecular conformation generation.
Besides Uni-Mol, the open-source models based on Uni-Mol include Uni-Mol+, Uni-Mol Tools, and Uni-Mol Docking V2. Uni-Mol+ is a model specifically designed for quantum chemical property prediction. By generating and optimizing initial three-dimensional conformations of two-dimensional molecular graphs, it achieves high-precision quantum chemical property predictions, significantly surpassing current state-of-the-art methods in multiple benchmark tests. Uni-Mol Tools is a set of user-friendly wrappers that support molecular property prediction, molecular representation, and other downstream tasks, providing a complete suite of practical tools based on the Uni-Mol model. Uni-Mol Docking V2 is the latest binding conformation prediction model, performing excellently in the PoseBusters benchmark test, with over 77% of predicted ligand binding conformations being accurate and RMSD values less than 2.0 Å. The generated prediction results are chemically accurate, avoiding issues such as chirality flipping and spatial conflicts. We will continue to open-source the latest foundational models and user-friendly toolkits and welcome contributions from the community, including models, tools, and documentation.
Uni-Mol Application Research Achievements
Currently, numerous researchers from various fields are using Uni-Mol for downstream application research, with articles published in top journals and international conferences such as Nature Communications, Journal of the American Chemical Society, Advanced Optical Materials, and NeurIPS. Uni-Mol+ will also be published in Nature Communications soon. Additionally, many research articles using Uni-Mol have been released in preprint form.
Future Development Plans for Uni-Mol
- Continually contributing to the community by improving the code, examples, and data, and consistently updating more downstream application cases.
- Open-sourcing more models and data, including the upcoming upgraded versions of Uni-Mol2 and Uni-Mol Docking.
- Iteratively updating versions of Uni-Mol Tools, adding new features, and supporting more data types, custom pre-training, and multi-type fine-tuning.
Uni-Mol in the DeepModeling Community
In the context of the current era of artificial intelligence and big data, Uni-Mol's integration into the DeepModeling open-source software community will provide the AI4S community with an efficient three-dimensional molecular pre-training framework, promoting collaborative research and development in molecular representation learning and pre-trained models.
Project Address
The Uni-Mol code is now open-source in the DeepModeling community. Feel free to use or join the project.
If you are interested in Uni-Mol or have new ideas, you are welcome to contact us via GitHub. If you wish to use Uni-Mol for research, please follow our website.