What Can DP + ABACUS Do Too? | Exploring Nuclear Quantum Effects in Electrochemical Reactions with Machine Learning and Developing Computational Methods
Recently, the research group led by Researcher Xu Shenzhen from the School of Materials Science and Engineering at Peking University collaborated with the Beijing Academy of Intelligent Sciences (AISI) and Deep Potential Technology (DP Technology). They employed the deep potential method[1] to study the nuclear quantum effects in proton-coupled electron transfer during electrochemical reactions and develop computational methods. Notably, all first-principles calculations in this study were carried out using the domestic first-principles software ABACUS [2]. The relevant research findings were published in Nature Communications under the title "Probing Nuclear Quantum Effects in Electrocatalysis via a Machine-Learning Enhanced Grand Canonical Constant Potential Approach" [3]. Doctoral students Sun Menglin, Jin Bin, and Yang Xiaolong are the co-first authors, and Researcher Xu Shenzhen is the corresponding author.
Research Background
Electrocatalytic systems can efficiently and cleanly convert electrical energy into chemical energy, making them one of the widely concerned fields in new energy technologies. They cover various scenarios such as water electrolysis, CO₂ reduction, and nitrogen reduction. The thermodynamic and kinetic properties of the elementary step of proton-coupled electron transfer (PCET) on the surface of electrode or catalyst materials, which is a crucial step in energy conversion, are key factors influencing performance indicators of electrocatalytic systems, such as product formation rate, energy conversion efficiency, and selectivity. They are also fundamental scientific issues that are the focus of experimental and theoretical electrochemical research.
Microscopic simulation of electrocatalytic systems is an important tool for studying the PCET mechanism. However, the electrochemical open system formed jointly by the electrode surface and the solution poses great challenges to computational simulation. How to consider the constant potential condition, sampling of the complex electrode-solution interface structure, and nuclear quantum effects (NQEs) of protons in a rigorous and efficient manner in the simulation has become a key problem to be solved urgently in the field of electrochemical theoretical calculation. For a typical elementary step of PCET on the electrode surface, such as A* + H+ + sol + e- ➜ AH (where "" represents the surface adsorption site), the traditional simulation method is to optimize the structures of the initial and final states of the reaction and then use the transition state search method to calculate the reaction energy barrier. The defects of this set of methods are as follows: (1) Only specific initial and final state structures are considered, and the reaction path is single, lacking statistical averaging; (2) In the simulation of the reaction path, the number of electrons in the surface system remains constant, which does not conform to the experimental condition of constant potential. Some post-treatment constant potential correction methods also cannot handle this problem strictly; (3) The nuclear quantum effects of hydrogen cannot be considered, while NQEs have been proven to have a significant impact on proton transfer kinetics in many other application scenarios (also at room temperature).
Based on the above problems, the research team developed a unified computational framework that can accurately handle NQEs under strict grand canonical constant potential conditions. Combined with the deep potential (DP) machine learning force field, it achieved sufficient sampling of electrochemical open systems. The DP force field can maintain an accuracy comparable to that of density functional theory (DFT) while greatly improving the computational efficiency, making it possible to conduct sufficient statistical sampling of the complex configurations of the electrode-solution interface in the reaction path at an affordable computational cost.
Research Results
This work is based on a rigorous statistical physics model. It uses the grand canonical hybrid Monte Carlo (GC-HMC) algorithm and the path integral Monte Carlo (PIMC) method, combined with the DP machine learning force field, to simulate the nuclear quantum effects of protons under precisely constant potential conditions. The workflow of this GC-(PI)HMC is shown in Figure 1a. In addition, based on the traditional DP machine learning potential function that takes atomic positions as inputs, the research team introduced a new input parameter degree of freedom: the total number of electrons Ne in the interface model system. Figure 1b shows the construction framework and training workflow of the DP-Ne machine learning force field used in this study. In order to calculate the work function W of the instantaneously sampled configuration in the extended space [R, Ne], an additional output term
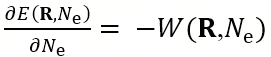
was introduced. For effective grand canonical ensemble sampling, it is necessary to satisfy:

represents the external electrochemical potential in equilibrium with the system and is a thermodynamic macroscopic quantity. The updated DP-Ne force field is helpful for sampling grand canonical systems with variable electron numbers.

Figure 1. a GC-(PI)HMC method workflow. The trial steps of different types of degrees of freedom are selected from PIMC, centroid atomic coordinates (R), and the number of electrons Ne using the random variable ξ according to a preset ratio. b The construction framework and training workflow of the DP-Ne force field used in this study. The added new degree of freedom - the total number of electrons Ne in the interface system is used as an input parameter for the fitting network.
The research team applied the new method to the hydrogen evolution reaction (HER) on the surface of a Pt electrode - a classic electrocatalytic science problem scenario, focusing on the impact of nuclear quantum effects on the thermodynamics and kinetics of the reaction. Electrocatalytic HER involves two basic PCET processes: Volmer (H+sol + e- + * ➜ H*), Heyrovsky (H+sol + e- + H* ➜ H2), and a non-electrochemical Tafel step (H* + H* ➜ H₂). After considering NQEs in the free energy calculations of the Volmer and Heyrovsky PCET steps, the obtained activation free energy results decreased by 0.1 - 0.15 eV (see Figure 2d), indicating that traditional electrocatalytic calculation models that do not consider NQEs may underestimate the PCET reaction rate at room temperature by dozens of times. The research results also provide a clear physical picture of proton tunneling when overcoming the energy barrier in the PCET path. The characteristic is that the position of the quantized proton has obvious uncertainty, reflecting the inherent quantum nature of proton transfer (see Figure 2e, 2f).
Figure 2. a Work function and b number of electrons fluctuations with the number of MC steps in the Volmer reaction. c The change process of the total number of electrons in the interface system during the reaction paths of Volmer and Heyrovsky reactions under classical conditions. d Free energy diagrams of Volmer and Heyrovsky steps under classical and quantum conditions. e Reaction coordinate (RC) distributions of ring-polymer beads in the path integral simulations of Volmer and Heyrovsky reactions. f Schematic diagrams of the structures of the beads when the transferred proton or hydrogen atom is considered with NQEs.
Summary
In this work, the researchers developed a GC-(PI)HMC computational framework that can explicitly handle NQEs under precisely constant potential conditions. With the assistance of a machine learning force field suitable for electrochemical conditions, it can accurately describe the PCET mechanism in electrochemistry. The work also reveals that NQEs have a non-negligible impact on HER on the Pt surface at room temperature. The quantum properties of the transferred protons are conducive to particles tunneling through classical potential barriers in the PCET path, thus significantly reducing the activation free energy compared with classical simulations. This discovery provides new physical insights into how protons overcome kinetic barriers during transfer.
This study not only provides new tools and methods for the theoretical research of electrocatalytic HER but also offers new research means for PCET reactions in a wider range of energy conversion processes. The computational framework developed by the research team is expected to become an important tool for future electrochemical simulations and provide theoretical support for the development of green energy technologies.
References
[1] Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 120, 143001 (2018).
[2] Chen, M., Guo, G.-C. & He, L. Systematically Improvable Optimized Atomic Basis Sets for Ab Initio Calculations. J. Phys.: Condens. Matter 22, 445501 (2010).
[3] Sun, M., Jin, B., Yang, X. & Xu, S. Probing Nuclear Quantum Effects in Electrocatalysis via a Machine-Learning Enhanced Grand Canonical Constant Potential Approach. Nature Communications 16, 3600 (2025)