What Can DP Do too? | Instant Electrochemical Potential Fluctuations Strongly Impact PCET Rate Constants in Constant-Potential Simulations
A research group led by Prof. Xu Shenzhen from the School of Materials Science and Engineering, Peking University, published the paper Electrochemical Potential Fluctuation Matters in Rate Constant Calculations for Proton-Coupled Electron Transfer in Journal of Chemical Theory and Computation. The study systematically compared two mainstream constant-potential molecular dynamics (MD) methods for predicting electrochemical reaction rates. Taking the Volmer step of the hydrogen evolution reaction (HER) on Pt(111) as the model system and adopting the Bennett-Chandler method, the team found that the rate constants calculated by the two approaches differ by approximately four times. The rigorous method allowing natural work function fluctuations (CPwfluc.) yields a rate constant around four times higher than the method fixing instantaneous work functions via electron number iteration (CPw/ofluc.).
Research Background:Challenges of Constant-Potential Electrochemical Simulations
Most core steps of electrochemical reactions such as hydrogen evolution, oxygen reduction and CO₂ reduction belong to proton-coupled electron transfer (PCET). Accurate rate prediction for PCET requires constant electrode potential simulations, which correspond to the grand canonical ensemble (GCE). In this ensemble, the system exchanges electrons with an external reservoir, leading to fluctuating electron numbers under fixed chemical potential.
Traditional first-principles calculations like density functional theory (DFT) are designed for the canonical ensemble with fixed electron counts. Direct grand canonical simulations are computationally expensive and technically difficult. Over the past decades, researchers have developed two types of constant-potential simulation strategies:
- CPwfluc. (with fluctuation): Complies strictly with GCE rules. It allows natural fluctuations of electron numbers, so the instantaneous work function oscillates around the set value.
- CPw/ofluc. (without fluctuation): Iteratively adjusts the electron number of each configuration to lock the work function at a target value and suppress inherent fluctuations.
CPwfluc. is theoretically rigorous in statistical physics, while CPw/ofluc. is widely used for its simple implementation. This work aims to quantify the discrepancy in reaction rates predicted by the two methods.
Comparision of Two Methods: Volmer Reaction Rate Constants
The research selected the Volmer step (H₃O⁺ + e⁻ + * → H* + H₂O), a key elementary reaction of HER on Pt(111) surfaces. The Bennett-Chandler method decomposes the rate constant into a thermodynamic activation term (probability density from the initial state to the transition state) and a dynamic recrossing term.
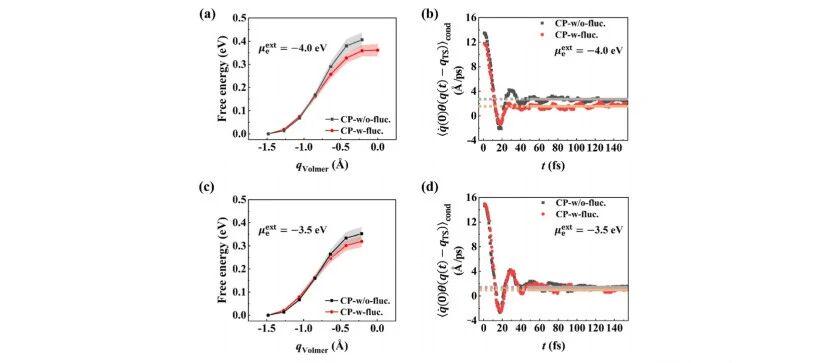
[Figure 1: (a,c) Free energy profiles along the defined reaction coordinate (RC); (b,d) Dynamic time-correlation function ⟨q̇(0)θ(q(t)−qTS)⟩cond at different electrochemical potentials calculated by CP-w-fluc. and CP-w/o-fluc. methods. ⟨⋅⟩cond refers to the conditional average under the constraint q(t=0)=qTS. Orange and gray guidelines mark the plateau values of time correlation functions for the two methods. Filled error bands in (a,c) represent standard errors derived from 10 independent mean force simulations.]
Under the same applied potential (μex = −3.5 eV vs. vacuum), the transition state probability density obtained by CPwfluc. reaches 1.35×10−5/Å, while CPw/ofluc. only gives 3.38×10−6/Å, a four-fold difference. A similar trend is observed at −4.0 eV.
For the dynamic recrossing term, the plateau values of flux-side time correlation functions from CPwfluc. are generally lower, indicating more significant recrossing effects captured by this method. Combined results of the two terms are listed in Table 1.

[Table 1: Reaction Rate Constants kBC at Different Electrochemical Potentials Calculated by CP-w/o-fluc. and CP-w-fluc. Methods]
In short, under identical applied potentials, the rate constant predicted by CPwfluc. is roughly four times that of CPw/ofluc.
In-depth Analysis: Physical Rigor vs. Operational Simplicity
The observed discrepancy reveals the essence of the grand canonical ensemble: fluctuations of electron numbers and potentials are microscopic manifestations of thermodynamic equilibrium between the system and the external environment. Artificially eliminating fluctuations replaces complete statistical averaging with maximum-probability approximation, making the partition function deviate from real GCE distribution.
Contrary to common perception, CPw/ofluc. is not more efficient. It requires repeated electron number iteration for convergence at every sampling step, resulting in heavier computational loads. In contrast, CPwfluc. evolves the total electron number $$N_e$$ as an independent dynamic sampling variable, featuring both stricter theories and higher computational efficiency.
The two methods can be unified within extended ensemble dynamics via the coupling strength Qμ:
- Qμ → 0: Corresponds to CPw/ofluc. with instantaneous electron equilibrium and no fluctuation;
- Moderate Qμ: Corresponds to CPwfluc. with dynamic fluctuations and accurate GCE sampling;
- Qμ → ∞: Corresponds to canonical ensemble sampling with fixed electron numbers.
DP + ABACUS: Enabling Accessible Accurate Constant-Potential Simulations
Conventional ab initio molecular dynamics (AIMD) makes CPwfluc. computationally costly. This work adopted the Deep Potential (DP) approach to address this problem.
All simulations were performed with neural network potentials trained by DP. The research team used the ABACUS first-principles software to generate DFT datasets and trained a DP- Ne machine learning potential via the automatic DP-GEN workflow. This model incorporates the total electron number Ne as well as its first and second partial derivatives. Leveraging numerical atomic orbital (NAO) basis sets, ABACUS efficiently handles large Pt/H₂O interface supercells containing hundreds of atoms. The DeePMD-kit trains deep learning force fields that precisely describe how atomic potential energy surfaces vary with electron numbers.
The final DP model achieves root-mean-square errors (RMSE) of 0.6 meV/atom for energy and 54 meV/Å for force, reaching first-principles precision. Based on this potential, the team conducted long-time sampling with 300,000 steps per trajectory for systematic method comparison. The DP-Ne model directly outputs ∂U/∂Ne and ∂2U/∂Ne2 via automatic differentiation, greatly accelerating electron number convergence in CPw/ofluc.
Conclusion
This study demonstrates that the selection of constant-potential simulation strategies is critical for reliable rate constant calculations of PCET elementary reactions. Instant electrochemical potential fluctuations are not negligible perturbations but core components of GCE statistical distribution. Neglecting such fluctuations with CPw/ofluc. will cause systematic deviations in rate constants — up to four times in the Volmer reaction investigated.
This work establishes an efficient constant-potential simulation framework based on DP machine learning force fields, enabling nanosecond-scale GCE MD sampling for large systems. It provides a new route for constant-potential simulations of complex electrochemical interfaces, such as electric double layers and dynamic evolution of electrocatalytic active sites.