What Can DP Do Too? | Deep Potential Reveals Cerium Segregation and Strengthening Mechanisms in Nanocrystalline Al-Ce Alloys
A research team led by Dr. Xue Hongtao and Dr. Chang Zhen from State Key Laboratory of Advanced Processing and Recycling of Non-ferrous Metals & Gansu Center for Materials Genome and Fundamental Structural Research, Lanzhou University of Technology, has published new findings in Journal of Materials Research and Technology. The paper titled Deep potential driven molecular dynamics simulations on tensile properties of nano-grained Al–Ce alloys: Mechanisms of Ce segregation and strengthening develops a high-precision deep potential (DP) for Al-Ce alloys and clarifies the microscale strengthening mechanism induced by grain boundary segregation of trace rare earth cerium (Ce) in nanocrystalline aluminum alloys.
Research Background
Molecular dynamics (MD) is a powerful atomistic simulation method, whose accuracy relies heavily on interatomic potentials. Ab initio molecular dynamics (AIMD) delivers high precision but suffers from exorbitant computational costs, limiting its application to small systems and short time scales. Deep potential (DP) effectively addresses this issue by achieving DFT-level accuracy at low computational overhead, enabling simulations of larger systems over longer durations.
Machine learning potentials have been widely adopted for various metals. However, due to the complex 4f electronic behavior of Ce, reliable potentials for rare-earth doped aluminum alloys (especially Al-Ce systems) remain scarce. Most existing studies focus on phase stability and thermodynamics, while systematic simulations on grain boundary segregation, dislocation motion and large-strain deformation in polycrystalline Al-Ce alloys are insufficient. Therefore, a high-fidelity DP model is urgently required to explore the strengthening mechanism of Ce-doped aluminum.
High-precision DP model: Enabling atomistic simulation of Al-Ce systems
The team adopted the DP-GEN active learning workflow to build the DP model for Al-Ce alloys, supporting atomistic analysis on the alloy’s tensile behaviors. Optimized thoroughly, this model accurately reproduces fundamental physical properties calculated via DFT, including energy-volume curves, equilibrium lattice parameters and elastic constants.
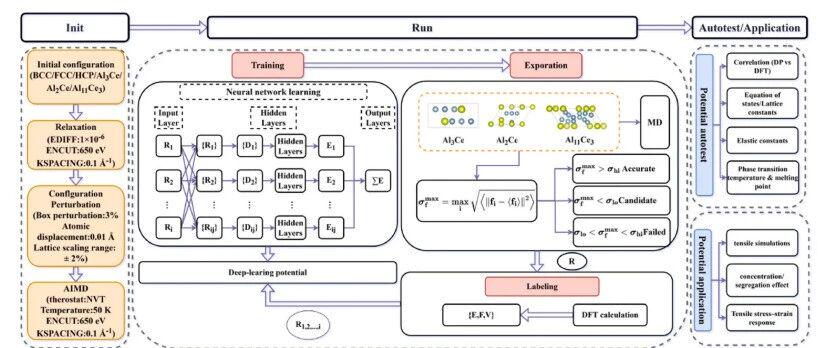
Figure 1: Flowchart of the development and application of machine learning potentials

Table 1: Equilibrium lattice constants (Å) of Al–Ce intermetallic phases (Al₁₁Ce₃, Al₃Ce, and Al₂Ce) obtained via DFT, DP and published literature
As shown in Table 1, the DP model predicts equilibrium lattice constants of Al₁₁Ce₃, Al₃Ce, Al₂Ce and other intermetallic phases with deviations below 1% compared with DFT results. Its predictions for elastic constants are also reliable, with most individual elastic constants deviating within 15%. The DP model also captures phase transition trends consistent with experimental observations. DP-based MD simulations show that the energy curves of α-Al₁₁Ce₃ and β-Al₁₁Ce₃ intersect at around 1300 K, which matches the experimental solid-state α→β phase transition temperature of 1279 K.
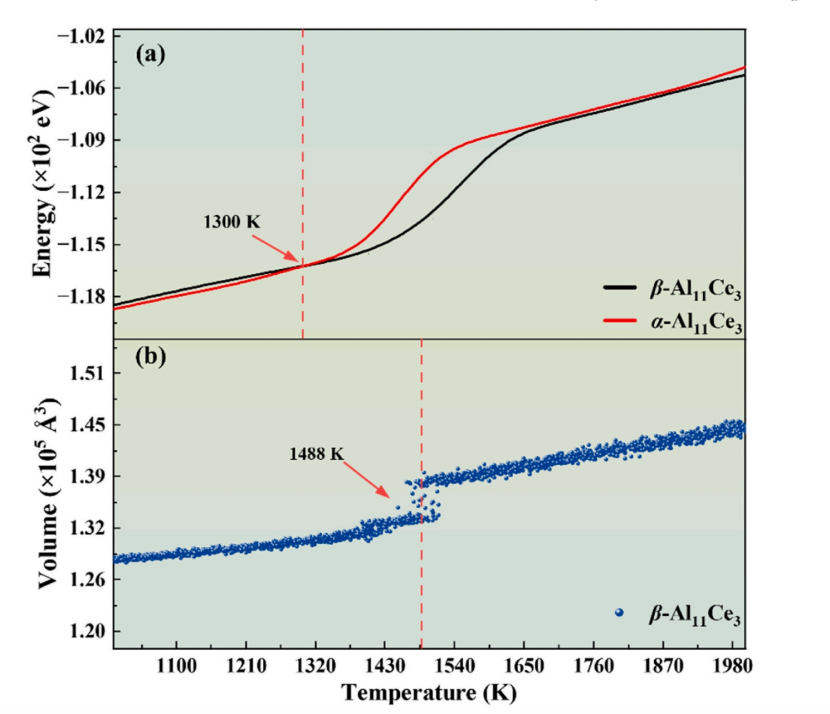
Figure 2: Temperature dependence of total energy and volume for Al₁₁Ce₃ polymorphs from DP-based molecular dynamics: (a) total energy-temperature curves of α-Al₁₁Ce₃ and β-Al₁₁Ce₃, (b) volume-temperature curve of β-Al₁₁Ce₃
Cerium segregation behavior: atomic-scale insights into grain boundary engineering
The researchers constructed nanocrystalline Al-Ce models with varying Ce atomic fractions (0, 0.010, 0.0154, 0.0195, 0.021, 0.027 at.%). Using a combined Monte Carlo (MC) and MD annealing scheme, they confirmed that Ce atoms tend to segregate and accumulate at grain boundaries.
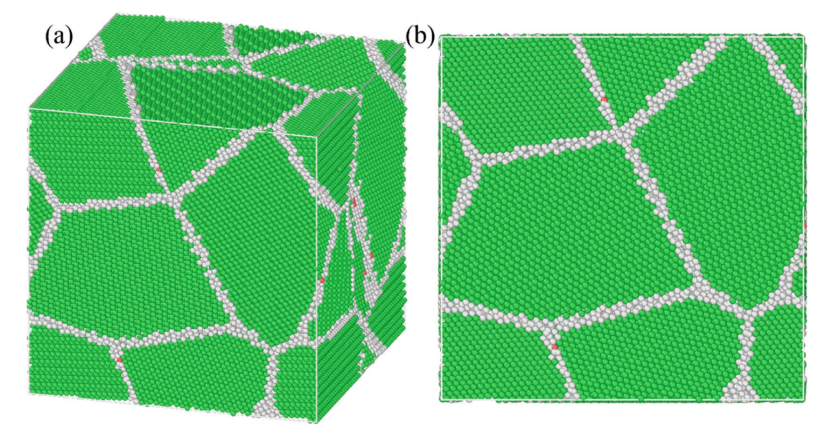
Figure 3: Atomic configurations of the Al–Ce models: (a) 3D view, (b) front view. Green: FCC grains; gray/red: grain boundaries and defects
After MC/MD annealing, the relative increase of Ce concentration at grain boundaries for samples with 0.010–0.027 at.% Ce is 27.65%, 85.01%, 3.84%, 36.33% and 13.84% respectively. The minimum increment occurs at 0.0195 at.% Ce, corresponding to the peak yield strength of the alloy.
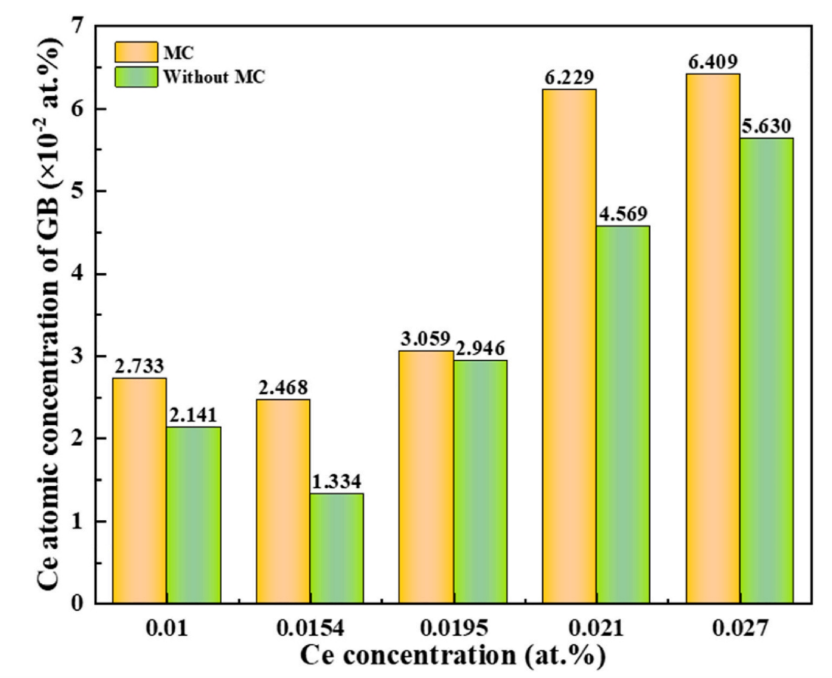
Figure 4: Fraction of Ce atoms at grain boundaries for different Ce contents, before and after MC equilibration at 300 K
Tensile Properties: Non-monotonic Variation Rule
All samples exhibit typical tensile characteristics of nanocrystalline metals, including a linear elastic stage, peak stress and plastic deformation with random fluctuations. Plastic deformation is mainly governed by dislocation movement and grain boundary sliding.
The yield strength presents a non-monotonic trend with rising Ce content (increasing first and then decreasing). All Al-Ce alloys possess higher yield strength than pure Al. Samples treated with MC/MD annealing always show better mechanical performance than those without annealing. For the 0.0195 at.% Ce sample, the yield strength rises from 1.846 GPa (without MC annealing) to 1.892 GPa (with MC annealing), a 2.5% improvement.

Figure 5: Tensile response of nanocrystalline Al–Ce alloys at different Ce contents: (a) stress-strain curves with MC/MD annealing; (b) stress-strain curves without MC annealing; (c) yield strength vs. Ce content
Strengthening Mechanism: Synergy of Dislocations and Stacking Faults
From 0.010 at.% to 0.0195 at.% Ce, the dislocation density near the yield point and the number of HCP-structured atoms gradually increase, followed by a decline at 0.027 at.% Ce. This variation is highly consistent with the trend of yield strength.
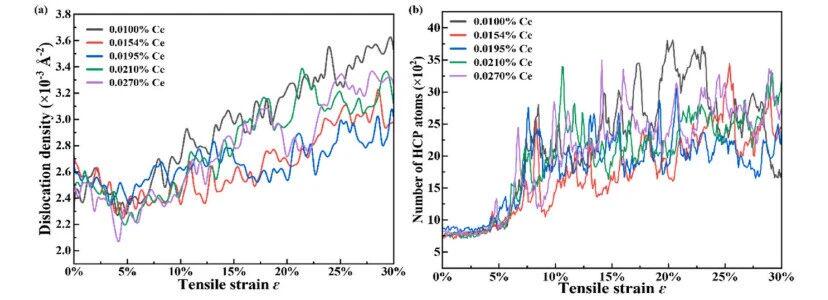
Figure 6: Dislocation density and HCP atoms of nanocrystalline Al–Ce alloys with different Ce contents: (a) dislocation density, (b) number of HCP atoms
At zero strain, dislocations are primarily distributed at grain boundaries and adjacent regions. Samples with higher yield strength have higher initial dislocation density at grain boundaries. For the 0.0195 at.% Ce sample, prominent activation of grain boundary dislocations and intensive dislocation interaction/tangling inside grains are observed.
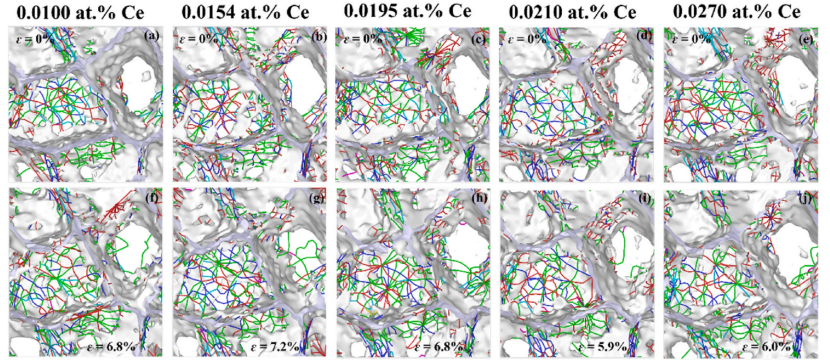
Figure 7: Dislocation networks in nanocrystalline Al–Ce alloys with different Ce contents: initial configurations at zero strain and configurations at the onset of yielding
Stacking faults preferentially nucleate at grain boundaries and triple junctions and then extend into grain interiors. At a tensile strain of approximately 10%, red HCP atoms emerge around grain boundaries and triple junctions, forming local stacking faults. As the strain increases to 18%–22%, stacking fault bands grow in quantity and length and interact with the grain boundary network.

Figure 8: Atomic snapshots of polycrystalline Al–Ce alloys with different Ce contents at selected tensile strains. Green: FCC grain interiors; gray: grain boundaries; red: HCP atoms
Conclusions and Outlook
Based on the high-precision DP model, this study systematically reveals the roles of Ce segregation and MC/MD annealing in strengthening nanocrystalline Al-Ce alloys. The yield strength reaches the maximum at 0.0195 at.% Ce, which is close to the commonly reported experimental value (~0.02 at.%) with a relative deviation of merely 2.5%. Compared with pure Al, the peak yield strength increases by 7.51%.
These findings deepen the understanding of the strengthening behavior of Al-Ce alloys and provide valuable theoretical guidance for the design and fabrication of high-performance aluminum alloy materials.
References
[1] Chang Z, Feng L, Xue HT, et al. Deep-learning potential molecular dynamics study on nanopolycrystalline Al-Er alloys: effects of Er concentration, grain boundary segregation, and grain size on plastic deformation. Journal of Chemical Information and Modeling, 2024;64:3282-93. https://doi.org/10.1021/acs.jcim.4c00008
[2] Xue HT, Chang Z, Li J, et al. Molecular dynamics simulations of the shear and tensile mechanical properties of rare-earth metal erbium based on deep-learning potential. Materials Today Communications, 2024;41:110485. https://doi.org/10.1016/j.mtcomm.2024.110485
[3] Li J, Xue HT, Zhang ZJ, et al. Deep-learning potential molecular dynamics simulations of the structural and physical properties of rare-earth alloys. Chinese Rare Earths, 2025;46(6):38-47. https://doi.org/10.3724/S1004-0277.202506005
[4] Xue HT, Li J, Chang Z, et al. Deep-learning potential molecular dynamics simulations of the structural and physical properties of rare-earth metal scandium. Computational Materials Science, 2024;242:113072. https://doi.org/10.1016/j.commatsci.2024.113072
Author Profile
Xue Hongtao, Associate Researcher and Doctoral Supervisor. He is a Gansu Provincial Science and Technology Commissioner (2026), a Visiting Scholar of the National "Western Light" Program (2023), a recipient of the Gansu Provincial Outstanding Youth Fund (2020) and Excellent Doctoral Dissertation Award (2017), and a Visiting Scholar at the University of Southampton, UK (2015). He is also an expert of Gansu Provincial Science and Technology Expert Database and Gansu Provincial AI Expert Think Tank.
His research focuses on multi-scale material simulations including machine learning-assisted first-principles calculations, molecular dynamics and Monte Carlo simulations. He devotes to structural design and performance regulation of metallic structural materials, thin-film solar cell materials and all-solid-state lithium-sulfur battery materials. He has presided over 2 national projects, 3 provincial projects, 2 departmental projects, 3 State Key Laboratory projects and 4 technical service projects. He has published over 100 SCI/EI-indexed papers in journals such as Acta Materialia and Materials Today. He serves as a youth editorial board member of Metals Advances and a reviewer for more than 10 top international journals including Nature Communications and Acta Materialia.