What Can Uni-Mol Do too? | Revealing the Critical Role of Molecular Conformations in AI Prediction Performance
Conformation, which refers to the different atomic arrangements a molecule can adopt in three-dimensional space, is one of the core factors determining a molecule's physicochemical properties. However, in current mainstream molecular machine learning modeling practices, conformational information is often ignored or simplified. Most prediction tasks still rely on two-dimensional structure representations, and there is even inconsistency in using the most stable conformational form. The lack of conformational information has become a significant factor limiting model prediction accuracy and also exposes the shortcomings of existing molecular representation methods in handling conformation-sensitive tasks.
Recently, Yu Hamakawa et al. published a study titled "Understanding Conformation Importance in Data-Driven Property Prediction Models" in the Journal of Chemical Information and Modeling. This research systematically evaluates the value of conformational information in molecular modeling and empirically compares the performance of mainstream models, including Uni-Mol, across multiple datasets. The study shows that by fully modeling conformational information, AI can significantly improve performance in multiple molecular property prediction tasks, with Uni-Mol demonstrating exceptional advantages in particular.
Dataset Introduction: Four Types of Task Scenarios with Conformational Diversity
To comprehensively investigate the impact of conformational information on prediction performance, the authors constructed and selected four representative datasets covering different task types, from theoretical property calculations to experimentally annotated results (see Figure 1):
- PQC (PubChemQC) Dataset: This dataset has the strongest conformational dependence in the study, containing over 90,000 small molecules annotated with six quantum chemical properties such as dipole moment, orbital energy levels, and electron affinity. It provides "true conformations" obtained from density functional theory (DFT) calculations as inputs, serving as an ideal testbed for evaluating the real performance contribution of conformational information.
- MP (Melting Point) Dataset: Compiled from experimental literature, it includes melting point information for 195 organic molecules. Since melting point is not a strongly conformation-sensitive property and conformational information is unknown, this dataset is more suitable for evaluating model performance and generalization ability under "no true conformation" conditions.
- APTC-1 and APTC-2 Datasets: Composed of small-sample catalytic reaction prediction tasks, they are used to model enantioselectivity in asymmetric catalytic reactions. These two datasets contain 88 and 40 samples, respectively, with unknown conformational information, but conformation plays a critical role in these tasks, making them important scenarios for testing a model's ability to "handle missing conformations."
Through this combination of four datasets, the study constructs a complete experimental matrix ranging from "with true conformation" to "without true conformation," from "large data volume" to "small samples," and from "weakly conformation-sensitive" to "strongly conformation-sensitive." This lays the foundation for systematically analyzing the mechanistic differences and performance boundaries of models in handling conformational information.
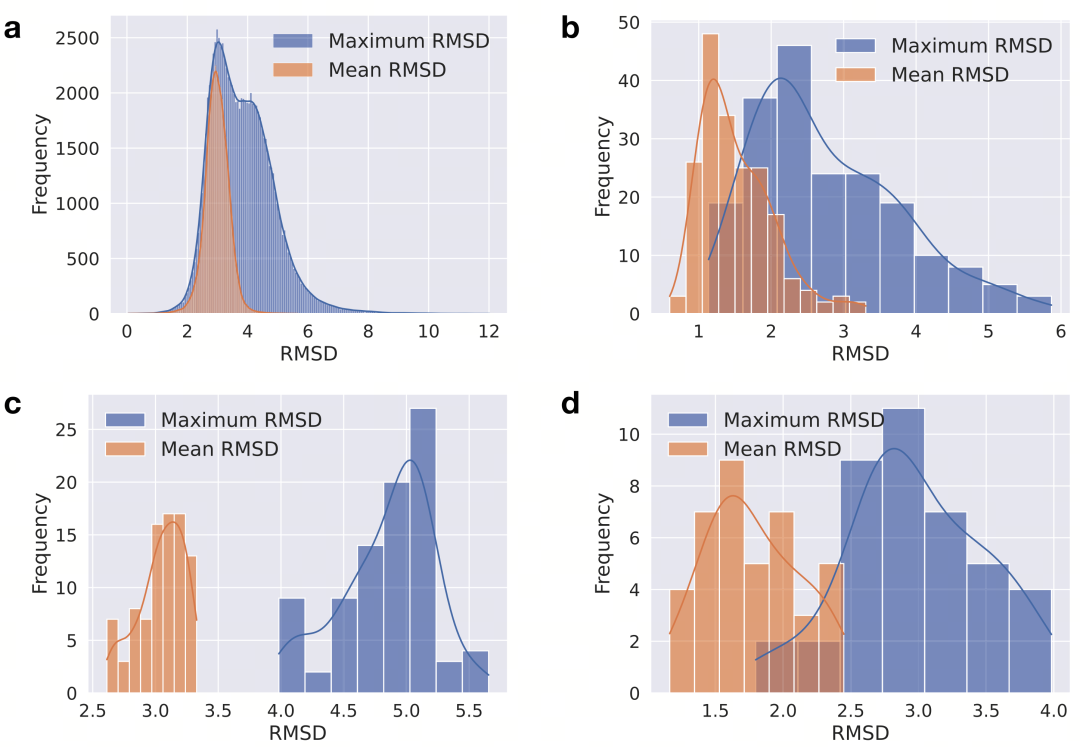
Figure 1. Conformational diversity of the four datasets. a-d show the distributions of the maximum and average atomic RMSD (unit: Å) among different conformations in the PQC, MP, APTC-1, and APTC-2 datasets, presented in blue and orange bar charts, respectively. Statistical data were obtained through exhaustive pairwise comparisons of all conformation pairs. RMSD calculations included hydrogen atoms and were optimized by rotation and translation to minimize RMSD values.
Dataset Modeling Process
Before detailing the models' performance across various tasks, the study uses the PQC dataset as a core case to construct a systematic modeling process (see Figure 2). This dataset consists of 100,000 molecules from PubChemQC, all structurally optimized using the PM6 Hamiltonian and annotated with six physicochemical properties: dipole moment (Debye), HOMO/LUMO energy levels (eV), energy gap (eV), total energy (eV), and enthalpy (Hartree). To ensure data balance and conformational diversity, the study restricts molecules to those containing only H, C, O, and N elements, with a molecular weight not exceeding 500, and preferentially selects molecular samples with more rotatable bonds.
In descriptor construction, the study calculates four types of three-dimensional descriptors (MOE, Pmapper, 3D-MoRSE, and MBTR) and one two-dimensional fingerprint (ECFP4). For multiple conformations of each molecule, six aggregation strategies (such as mean, Boltzmann weighting, minimum energy, and non-aggregation) are used to summarize the 3D descriptors. Meanwhile, descriptors of "true conformations" are retained as a control group. This processed representation data provides a unified and controllable input basis for subsequent performance comparisons of different models in "conformational sensitivity" analyses.
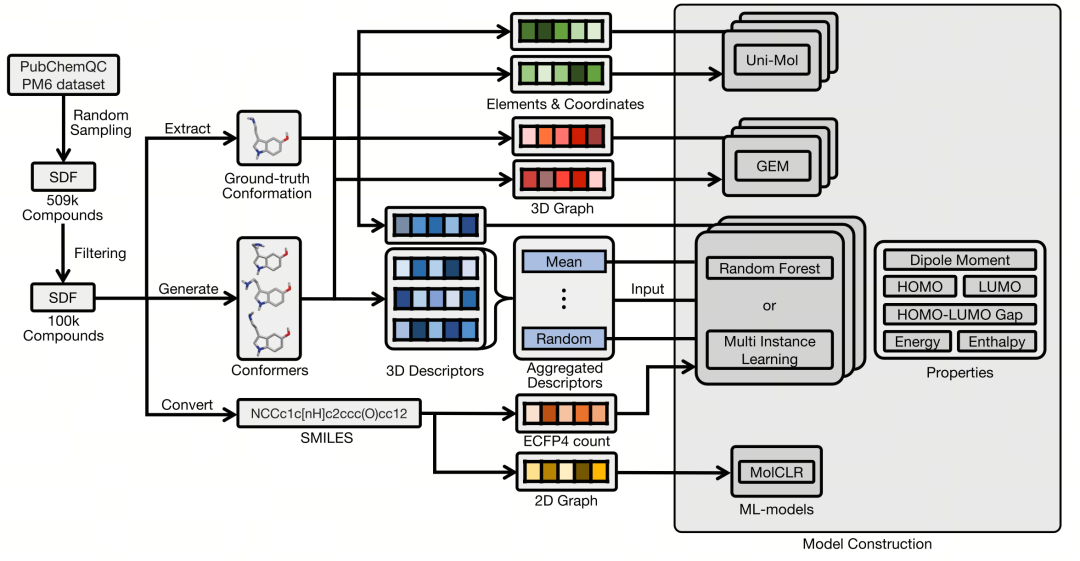
Figure 2 shows the modeling process using the PQC dataset. The PQC dataset contains the atomic coordinate information of compounds optimized by the PM6 Hamiltonian, as well as the labeled values of the following six properties: dipole moment (unit: Debye), HOMO energy level (unit: eV), LUMO energy level (unit: eV), HOMO - LUMO energy gap (unit: eV), total energy (unit: eV), and enthalpy (unit: Hartree). The study randomly selected molecular samples from a sub - dataset that contains only four elements, hydrogen, carbon, oxygen, and nitrogen, and has a molecular weight not exceeding 500. These molecules were sorted according to the number of rotatable bonds, and finally, the top 100,000 molecules with the largest number of rotatable bonds were selected for modeling. For multiple conformations of each molecule, the study calculated four molecular descriptors respectively: three types of three - dimensional descriptors (MOE, Pmapper, 3D - MoRSE, and MBTR) and one two - dimensional descriptor (ECFP4 count - type).
Comparison of Multi-Conformation Strategies: Does Quantity Equal Quality?
The authors address a fundamental yet critical question: For multiple conformations of the same molecule, does an increase in quantity necessarily lead to improved prediction accuracy? Using the PQC dataset as an example, the study examines the performance of various aggregation strategies (including mean, minimum energy, and Boltzmann weighting) at different numbers of conformations. As shown in Figure 3, the results indicate that for most models, the benefits of increasing the number of conformations are not significant, and there is even an issue of "information redundancy." Under the same conditions, however, Uni-Mol demonstrates high adaptability to the number of conformations, with its performance continuously improving and significantly surpassing traditional aggregation strategy models after using 10 conformations.
This phenomenon reveals an essential advantage of the Uni-Mol architecture: it can not only utilize information about "which conformation" is present but also understand the "conformational distribution" itself, thereby capturing spatial features beyond simple averaging or weighting strategies.
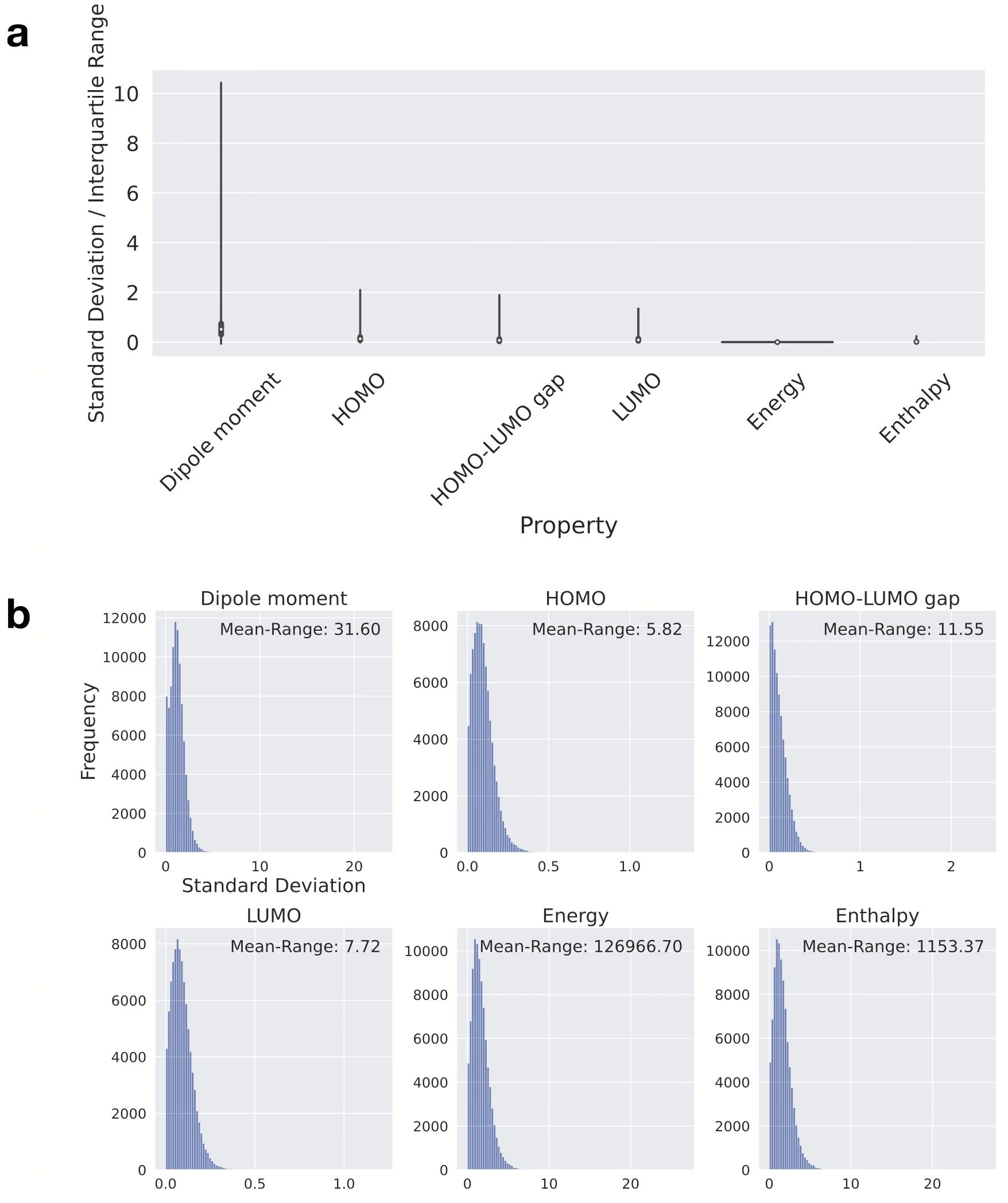
Figure 3 :Sensitivity analysis of molecular properties to conformational differences. a, Violin plot of the correlation between the standard deviation of six properties and conformational differences, standardized by the interquartile range of each property. Multiple conformations of each molecule were used to calculate the standard deviation of its properties, which were further standardized. b, Histogram of the original standard deviation before standardization. The range of change in the average value of each property is given in the figure. The unit of dipole moment is Debye, HOMO, LUMO and their energy gaps are in eV, and energy and enthalpy are in kcal/mol.
Accuracy Comparison: Uni-Mol's Strong Performance in Conformation-Sensitive Tasks
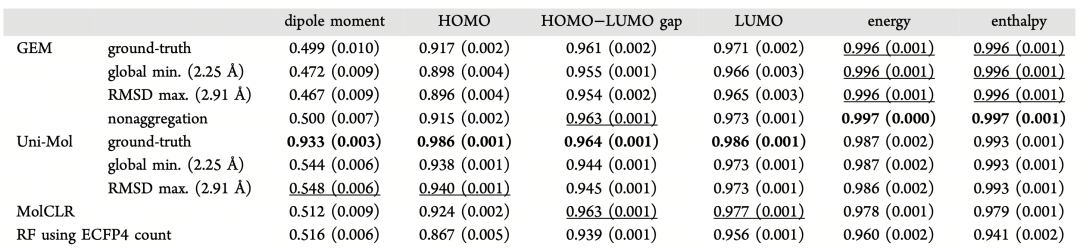
Table 1 shows the prediction performance of various methods on six physicochemical properties in the PQC dataset, including dipole moment, HOMO energy level, and electron affinity. When using "true conformation" inputs, Uni-Mol leads in performance for most properties. Notably, in dipole moment prediction, its R² value reaches 0.933, far higher than traditional ECFP fingerprints (0.516) and MOE 3D descriptors (0.498).
Even when using "non-true" conformations (such as minimum energy or RDKit default conformations), Uni-Mol still maintains significant predictive advantages. This indicates its robustness in handling conformational perturbations and good fault tolerance for situations with incomplete or inaccurate conformational information in practical applications.
Table 1 Comparison of prediction performance under different models and conformation input methods. The R² performance of six quantum property predictions on the PQC dataset for three types of deep models (MolCLR, GEM, Uni-Mol) is presented, using true conformations, minimum energy conformations, and maximum RMSD conformations, respectively.
Generalization Ability Validation: How Uni-Mol Handles Data Beyond the Chemical Space
While the ability to model conformations is important, a more practical challenge is the model's extrapolation capability for molecules with low similarity. Figure 4 shows a generalization experiment based on Tanimoto similarity classification: as the structural similarity between test molecules and the training set gradually decreases, the prediction performance of most traditional models (especially those based on two-dimensional fingerprints) declines rapidly. In contrast, Uni-Mol exhibits a slower and more stable decline, maintaining an R² value above 0.6 for the "least similar" molecular group.
This suggests that Uni-Mol has not merely learned the feature correlations existing in the training set but has mastered a deeper structural-property mapping rule. Its conformational understanding ability is not only applicable to known molecular systems but also has the potential to extend to unknown chemical spaces.
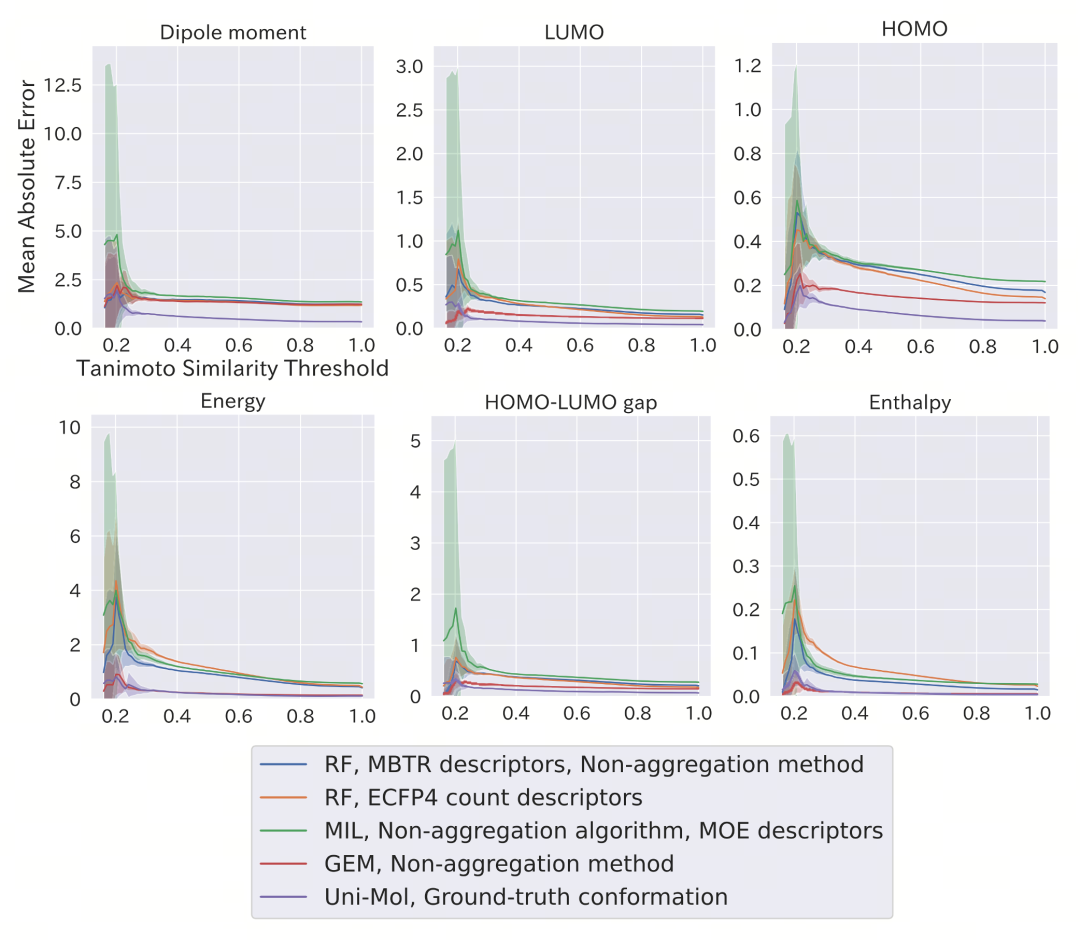
Figure 4: Robustness analysis of the model in chemical space extrapolation. When the maximum Tanimoto similarity between the test set and the training set is lower than a specific threshold, the changing trend of the prediction error (MAE) of each model. The horizontal axis represents the similarity threshold, the vertical axis represents the average MAE, and the shaded area represents the standard deviation. Uni-Mol still maintains a low error in the low-similarity region, indicating its strong extrapolation ability.
Modeling Capability in Conformation-Lacking Scenarios: Empirical Validation in Small Samples
When further exploring modeling capabilities in real-world tasks, the authors selected two small-sample datasets for enantioselectivity prediction—APTC-1 and APTC-2—to empirically compare various modeling strategies. Unlike the previously mentioned quantum chemistry tasks with known conformations, these two datasets not only have very few samples (88 and 40 reactions, respectively) but also do not provide "true conformations," making them closer to the challenges of scarce data and structural uncertainty in real scientific research.
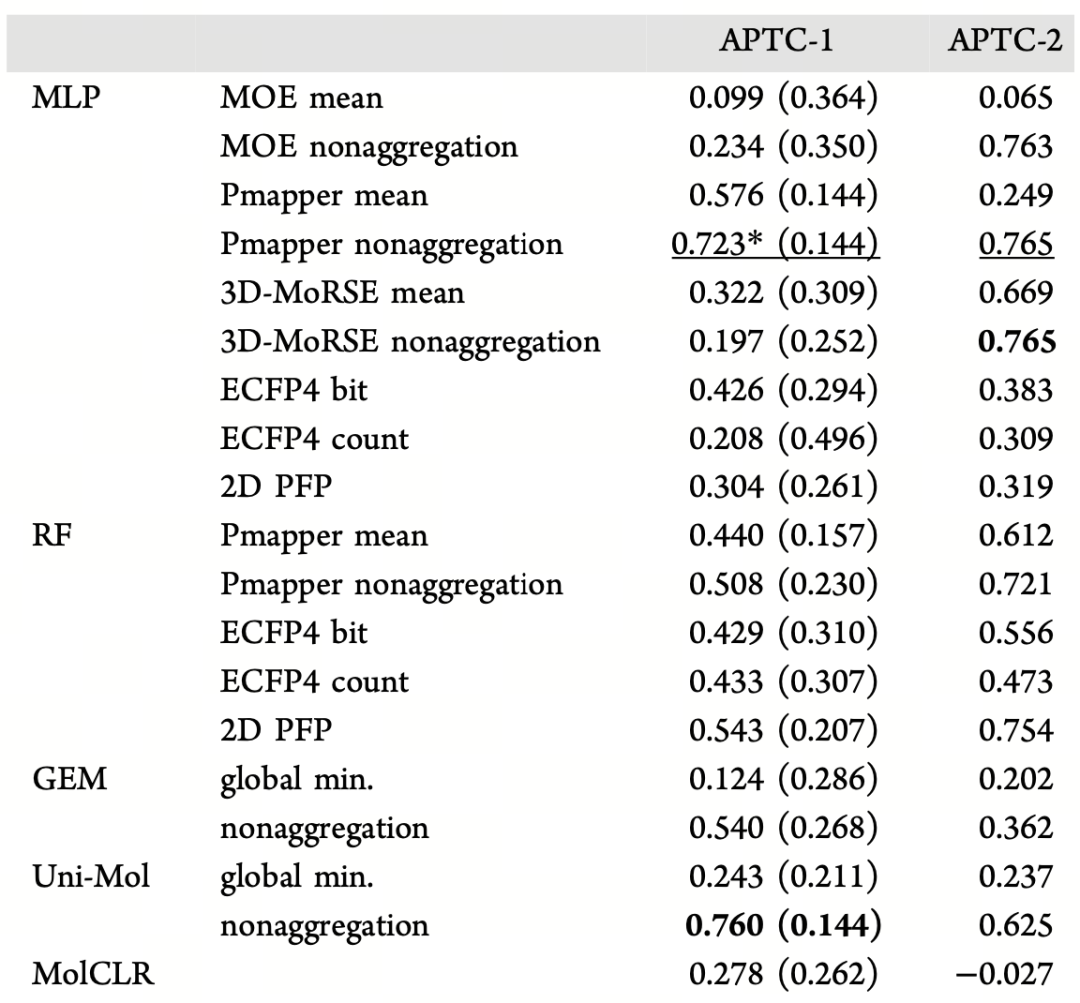
As shown in Table 2, when using non-aggregation strategies to input conformational information, Uni-Mol achieves an R² of 0.760 in the APTC-1 task, significantly higher than other methods such as MLP (0.723), GEM (0.540), and MolCLR (0.278). In the APTC-2 dataset, its performance remains robust, with an R² as high as 0.625. Notably, compared to Uni-Mol using single-conformation (global min) inputs (R² values of only 0.243 and 0.237), the non-aggregation approach fully unleashes the potential of multi-conformational information, leading to a significant improvement.
Table 2 Comparison of the average accuracy (R²) of different machine learning models in predicting enantioselectivity (ΔΔG‡) in the APTC-1 (25 test sets) and APTC-2 (40 test points) datasets
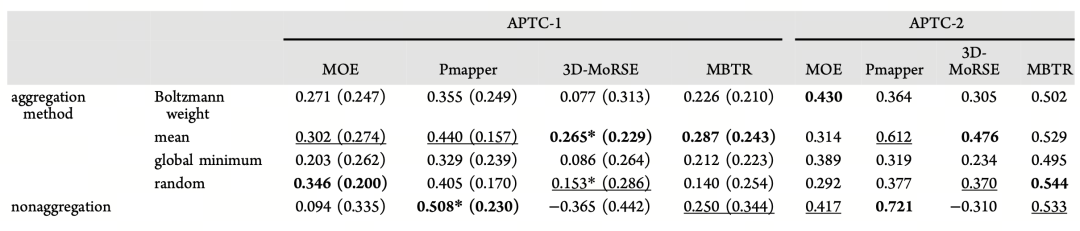
Meanwhile, as shown in Table 3, even for traditional random forest models, non-aggregation strategies generally outperformed aggregation strategies when using 3D descriptors such as MOE or Pmapper. For example, in APTC-1, the non-aggregation strategy with Pmapper achieved an R² of 0.508, representing the best performance under this descriptor. This trend further validates a core perspective: in real-world tasks lacking true conformations, preserving conformational diversity holds more modeling value than single-state aggregation.
Table 3 Average accuracy (R²) of the random forest (RF) model in predicting enantioselectivity (ΔΔG‡) on the APTC-1 (25 test sets) and APTC-2 (40 test points) datasets
Conclusion: Uni-Mol's Systematic Response to Conformation-Sensitive Problems
This study systematically reveals the importance of conformational information in molecular property prediction tasks through comparisons across multiple datasets, modeling methods, and conformational processing strategies, particularly in scenarios involving small samples, unknown conformations, or highly structure-sensitive tasks. As an end-to-end three-dimensional molecular modeling framework, Uni-Mol not only demonstrates strong adaptability in conformational analysis and representation strategies but also deeply integrates spatial invariance and structure-aware capabilities through SE(3)-equivariant structures, significantly enhancing model stability and generalization.
The value of Uni-Mol extends beyond performance metric improvements and has been validated in multiple practical research scenarios. In drug discovery, it is used for binding site screening, active molecule generation, and conformation-sensitive property modeling. In materials science, its structure-aware capabilities are applied to cutting-edge topics such as molecular crystal prediction and band structure analysis. As a highly modular and extensible three-dimensional modeling platform, Uni-Mol is gradually becoming an important foundation in the field of AI chemistry.