Efficiently Trained Deep Learning Potential for Graphane
Siddarth K. Achar, Linfeng Zhang, J. Karl Johnson
The Journal of Physical Chemistry C, 2021, 125 (27), 14874–14882.
DOI: 10/gmfwwb
Cormorant: Covariant Molecular Neural Networks
Brandon Anderson, Truong-Son Hy, Risi Kondor
Advances in Neural Information Processing Systems 32 (Nips 2019), 2019, 32.
Optimization and Validation of a Deep Learning CuZr Atomistic Potential: Robust Applications for Crystalline and Amorphous Phases with near-DFT Accuracy
Christopher M. Andolina, Philip Williamson, Wissam A. Saidi
Journal of Chemical Physics, 2020, 152 (15).
DOI: 10.1063/5.0005347
Robust, Multi-Length-Scale, Machine Learning Potential for Ag–Au Bimetallic Alloys from Clusters to Bulk Materials
Christopher M. Andolina, Marta Bon, Daniele Passerone, Wissam A. Saidi
The Journal of Physical Chemistry C, 2021.
DOI: 10/gmdj4k
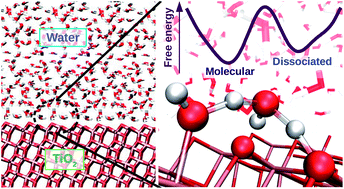
Free Energy of Proton Transfer at the Water-TiO2 Interface from Ab Initio Deep Potential Molecular Dynamics
Marcos F. Calegari Andrade, Hsin-Yu Ko, Linfeng Zhang, Roberto Car, Annabella Selloni
Chemical Science, 2020, 11 (9), 2335–2341.
DOI: 10.1039/c9sc05116c
Hydrogen Dynamics in Supercritical Water Probed by Neutron Scattering and Computer Simulations
Carla Andreani, Giovanni Romanelli, Alexandra Parmentier, Roberto Senesi, Alexander Kolesnikov, Hsin-Yu Ko, Marcos F. Calegari Andrade, Roberto Car
Journal of Physical Chemistry Letters, 2020, 11 (21), 9461–9467.
DOI: 10.1021/acs.jpclett.0c02547
Active Learning Accelerates Ab Initio Molecular Dynamics on Pericyclic Reactive Energy Surfaces
Shi Jun Ang, Wujie Wang, Daniel Schwalbe-Koda, Simon Axelrod, Rafael Gomez-Bombarelli
2020.
Active Learning Accelerates Ab Initio Molecular Dynamics on Reactive Energy Surfaces
Shi Jun Ang, Wujie Wang, Daniel Schwalbe-Koda, Simon Axelrod, Rafael Gómez-Bombarelli
Chem, 2021, 7 (3), 738–751.
DOI: 10/gmgdj2
Embedding Quantum Statistical Excitations in a Classical Force Field
Susan R. Atlas
Journal of Physical Chemistry A, 2021, 125 (17), 3760–3775.
DOI: 10.1021/acs.jpca.1c00164
Deep Machine Learning Interatomic Potential for Liquid Silica
I. A. Balyakin, S. Rempel, R. E. Ryltsev, A. A. Rempel
Physical Review E, 2020, 102 (5), 052125.
DOI: 10.1103/PhysRevE.102.052125
Machine-Learning-Based Interatomic Potential for Phonon Transport in Perfect Crystalline Si and Crystalline Si with Vacancies
Hasan Banaei, Ruiqiang Guo, Amirreza Hashemi, Sangyeop Lee
Physical Review Materials, 2019, 3 (7), 074603.
DOI: 10.1103/PhysRevMaterials.3.074603
Structure Motif-Centric Learning Framework for Inorganic Crystalline Systems
Huta R. Banjade, Sandro Hauri, Shanshan Zhang, Francesco Ricci, Weiyi Gong, Geoffroy Hautier, Slobodan Vucetic, Qimin Yan
Science Advances, 2021, 7 (17), eabf1754.
DOI: 10.1126/sciadv.abf1754
Voxelized Atomic Structure Potentials: Predicting Atomic Forces with the Accuracy of Quantum Mechanics Using Convolutional Neural Networks
Matthew C. Barry, Kristopher E. Wise, Surya R. Kalidindi, Satish Kumar
Journal of Physical Chemistry Letters, 2020, 11 (21), 9093–9099.
DOI: 10.1021/acs.jpclett.0c02271
Machine Learning a General-Purpose Interatomic Potential for Silicon
Albert P. Bartók, James Kermode, Noam Bernstein, Gábor Csányi
Physical Review X, 2018, 8 (4), 041048.
DOI: 10.1103/PhysRevX.8.041048
Machine Learning for Multi-Fidelity Scale Bridging and Dynamical Simulations of Materials
R Batra, S Sankaranarayanan - Journal of Physics: Materials, undefined 2020
iopscience.iop.org, 2020, 3, 31002.
DOI: 10.1088/2515-7639/ab8c2d
SE(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, Boris Kozinsky
2021.
De Novo Exploration and Self-Guided Learning of Potential-Energy Surfaces
Noam Bernstein, Gabor Csanyi, Volker L. Deringer
Npj Computational Materials, 2019, 5, 99.
DOI: 10.1038/s41524-019-0236-6
A Perspective on Inverse Design of Battery Interphases Using Multi-Scale Modelling, Experiments and Generative Deep Learning
Arghya Bhowmik, Ivano E. Castelli, Juan Maria Garcia-Lastra, Peter Bjorn Jorgensen, Ole Winther, Tejs Vegge
Energy Storage Materials, 2019, 21, 446–456.
DOI: 10.1016/j.ensm.2019.06.011
Efficient Sampling of Equilibrium States Using Boltzmann Generators
Jeremy Binagia, Sean Friedowitz, Kevin J Hou
, 6.
Efficient Global Structure Optimization with a Machine-Learned Surrogate Model
Malthe K. Bisbo, Bjørk Hammer
Physical Review Letters, 2020, 124 (8).
DOI: 10.1103/physrevlett.124.086102
Efficient Prediction of 3D Electron Densities Using Machine Learning
Mihail Bogojeski, Felix Brockherde, Leslie Vogt-Maranto, Li Li, Mark E. Tuckerman, Kieron Burke, Klaus-Robert Müller
2018.
Quantum Chemical Accuracy from Density Functional Approximations via Machine Learning
Mihail Bogojeski, Leslie Vogt-Maranto, Mark E. Tuckerman, Klaus-Robert Mueller, Kieron Burke
Nature Communications, 2020, 11 (1), 5223.
DOI: 10.1038/s41467-020-19093-1
Neural Networks-Based Variationally Enhanced Sampling
Luigi Bonati, Yue-Yu Zhang, Michele Parrinello
Proceedings of the National Academy of Sciences of the United States of America, 2019, 116 (36), 17641–17647.
DOI: 10.1073/pnas.1907975116
Silicon Liquid Structure and Crystal Nucleation from Ab Initio Deep Metadynamics
Luigi Bonati, Michele Parrinello
Physical review letters, 2018, 121 (26), 265701.
DOI: 10.1103/PhysRevLett.121.265701
Machine Learning in Nano-Scale Biomedical Engineering
Alexandros-Apostolos A. Boulogeorgos, Stylianos E. Trevlakis, Sotiris A. Tegos, Vasilis K. Papanikolaou, George K. Karagiannidis
2020.
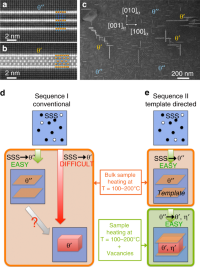
Transforming Solid-State Precipitates via Excess Vacancies
Laure Bourgeois, Yong Zhang, Zezhong Zhang, Yiqiang Chen, Nikhil Medhekar
Nature Communications, 2020, 11 (1), 1248.
DOI: 10.1038/s41467-020-15087-1
MB-Fit: Software Infrastructure for Data-Driven Many-Body Potential Energy Functions
Ethan Bull-Vulpe, Marc Riera, Andreas Goetz, Francesco Paesani
2021.
Deep-Learning Approach to First-Principles Transport Simulations
Marius Burkle, Umesha Perera, Florian Gimbert, Hisao Nakamura, Masaaki Kawata, Yoshihiro Asai
Physical Review Letters, 2021, 126 (17), 177701.
DOI: 10.1103/PhysRevLett.126.177701
Gaussian Approximation Potentials for Body-Centered-Cubic Transition Metals
J. Byggmastar, K. Nordlund, F. Djurabekova
Physical Review Materials, 2020, 4 (9), 093802.
DOI: 10.1103/PhysRevMaterials.4.093802
Machine-Learning Interatomic Potential for Radiation Damage and Defects in Tungsten
J. Byggmastar, A. Hamedani, K. Nordlund, F. Djurabekova
Physical Review B, 2019, 100 (14), 144105.
DOI: 10.1103/PhysRevB.100.144105
Structure of Disordered \${\textbackslash mathrm{\vphantom}}TiO\vphantom{}\vphantom{}_{2}\$ Phases from Ab Initio Based Deep Neural Network Simulations
Marcos F. Calegari Andrade, Annabella Selloni
Physical Review Materials, 2020, 4 (11), 113803.
DOI: 10/ghnhd5
Machine-Learning X-Ray Absorption Spectra to Quantitative Accuracy
Matthew R. Carbone, Mehmet Topsakal, Deyu Lu, Shinjae Yoo
Physical Review Letters, 2020, 124 (15), 156401.
DOI: 10.1103/PhysRevLett.124.156401
Computing RPA Adsorption Enthalpies by Machine Learning Thermodynamic Perturbation Theory
Bilal Chehaibou, Michael Badawi, Tomas Bucko, Timur Bazhirov, Dario Rocca
Journal of Chemical Theory and Computation, 2019, 15 (11), 6333–6342.
DOI: 10.1021/acs.jctc.9b00782
Topics in the Mathematical Design of Materials
X Chen, I Fonseca, M Ravnik, V Slastikov, C Zannoni
Philosophical transactions. Series A, Mathematical, physical, and engineering sciences, 2021, 379 (2201), 20200108.
DOI: 10.1098/rsta.2020.0108
Direct Prediction of Phonon Density of States with Euclidean Neural Networks
Z Chen, N Andrejevic, T Smidt, Z Ding, Q Xu - Advanced …, undefined 2021
Wiley Online Library, 2021, 8.
DOI: 10.1002/advs.202004214
Atomic Energies from a Convolutional Neural Network
Xin Chen, Mathias S. Jorgensen, Jun Li, Bjork Hammer
Journal of Chemical Theory and Computation, 2018, 14 (7), 3933–3942.
DOI: 10.1021/acs.jctc.8b00149
Competitive Effect of Disorder and Defects on Dynamic Structural Transformation of Compressed Gold
B Chen, Q Zeng, H Wang, D Kang, J Dai
arxiv.org, 2021.
DOI: arXiv:2006.13136
A Critical Review of Machine Learning of Energy Materials
Chi Chen, Yunxing Zuo, Weike Ye, Xiangguo Li, Zhi Deng, Shyue Ping Ong
Advanced Energy Materials, 2020, 10 (8), 1903242.
DOI: 10.1002/aenm.201903242
Machine Learning on Neutron and X-Ray Scattering
Z Chen, N Andrejevic, N Drucker, T Nguyen
arxiv.org.
DeePKS: A Comprehensive Data-Driven Approach toward Chemically Accurate Density Functional Theory
Yixiao Chen, Linfeng Zhang, Han Wang, E. Weinan
Journal of Chemical Theory and Computation, 2021, 17 (1), 170–181.
DOI: 10.1021/acs.jctc.0c00872
DeePKS-Kit: A Package for Developing Machine Learning-Based Chemically Accurate Energy and Density Functional Models
Y Chen, L Zhang, H Wang
arxiv.org, 2021.
Efficient Construction of Excited-State Hessian Matrices with Machine Learning Accelerated Multilayer Energy-Based Fragment Method
Wen-Kai Chen, Yaolong Zhang, Bin Jiang, Wei-Hai Fang, Ganglong Cui
Journal of Physical Chemistry A, 2020, 124 (27), 5684–5695.
DOI: 10.1021/acs.jpca.0c04117
Exploiting Machine Learning to Efficiently Predict Multidimensional Optical Spectra in Complex Environments
Michael S. Chen, Tim J. Zuehlsdorff, Tobias Morawietz, Christine M. Isborn, Thomas E. Markland
Journal of Physical Chemistry Letters, 2020, 11 (18), 7559–7568.
DOI: 10.1021/acs.jpclett.0c02168
Co-Segregation of Mg and Zn Atoms at the Planar Η1-Precipitate/Al Matrix Interface in an Aged Al–Zn–Mg Alloy
Bingqing Cheng, Xiaojun Zhao, Yong Zhang, Houwen Chen, Ian Polmear, Jian-Feng Nie
Scripta Materialia, 2020, 185, 51–55.
DOI: 10/gmgc5h
Deep-Learning Potential Method to Simulate Shear Viscosity of Liquid Aluminum at High Temperature and High Pressure by Molecular Dynamics
Yuqing Cheng, Han Wang, Shuaichuang Wang, Xingyu Gao, Qiong Li, Jun Fang, Hongzhou Song, Weidong Chu, Gongmu Zhang, Haifeng Song, Haifeng Liu
Aip Advances, 2021, 11 (1), 015043.
DOI: 10.1063/5.0036298
Gold Segregation Improves Electrocatalytic Activity of Icosahedron Au@Pt Nanocluster: Insights from Machine Learning
Dingming Chen, Zhuangzhuang Lai, Jiawei Zhang, Jianfu Chen, Peijun Hu, Haifeng Wang
Chinese Journal of Chemistry, 2021, n/a (n/a).
DOI: 10/gmfw5g
Regression Clustering for Improved Accuracy and Training Costs with Molecular-Orbital-Based Machine Learning
Lixue Cheng, Nikola B. Kovachki, Matthew Welborn, Thomas F. Miller
Journal of Chemical Theory and Computation, 2019, 15 (12), 6668–6677.
DOI: 10.1021/acs.jctc.9b00884
Ground State Energy Functional with Hartree-Fock Efficiency and Chemical Accuracy
Yixiao Chen, Linfeng Zhang, Han Wang, E. Weinan
Journal of Physical Chemistry A, 2020, 124 (35), 7155–7165.
DOI: 10.1021/acs.jpca.0c03886
A Universal Density Matrix Functional from Molecular Orbital-Based Machine Learning: Transferability across Organic Molecules
Lixue Cheng, Matthew Welborn, Anders S. Christensen, Thomas F. Miller
Journal of Chemical Physics, 2019, 150 (13), 131103.
DOI: 10.1063/1.5088393
Integrating Machine Learning with the Multilayer Energy-Based Fragment Method for Excited States of Large Systems
Wen-Kai Chen, Wei-Hai Fang, Ganglong Cui
Journal of Physical Chemistry Letters, 2019, 10 (24), 7836–7841.
DOI: 10.1021/acs.jpclett.9b03113
On the Representation of Solutions to Elliptic PDEs in Barron Spaces
Ziang Chen, Jianfeng Lu, Yulong Lu
2021.
TensorAlloy: An Automatic Atomistic Neural Network Program for Alloys
Xin Chen, Xing-Yu Gao, Ya-Fan Zhao, De-Ye Lin, Wei-Dong Chu, Hai-Feng Song
Computer Physics Communications, 2020, 250, 107057.
DOI: 10.1016/j.cpc.2019.107057
Unsupervised Machine Learning Methods for Polymer Nanocomposites Data via Molecular Dynamics Simulation
Zhudan Chen, Dazi Li, Haixiao Wan, Minghui Liu, Jun Liu
Molecular Simulation, 2020.
DOI: 10.1080/08927022.2020.1851028
Constructing Convex Energy Landscapes for Atomistic Structure Optimization
Siva Chiriki, Mads-Peter Christiansen, B. Hammer
Physical Review B, 2019, 100 (23), 235436.
DOI: 10.1103/PhysRevB.100.235436
Accurate Molecular Dynamics Enabled by Efficient Physically-Constrained Machine Learning Approaches
Stefan Chmiela, Huziel E. Sauceda, Alexandre Tkatchenko, Klaus-Robert Müller
2020, 968, 129–154.
DOI: 10/gmgfsq
Towards Exact Molecular Dynamics Simulations with Machine-Learned Force Fields
Stefan Chmiela, Huziel E. Sauceda, Klaus-Robert Mueller, Alexandre Tkatchenko
Nature Communications, 2018, 9, 3887.
DOI: 10.1038/s41467-018-06169-2
sGDML: Constructing Accurate and Data Efficient Molecular Force Fields Using Machine Learning
Stefan Chmiela, Huziel E. Sauceda, Igor Poltavsky, Klaus-Robert Mueller, Alexandre Tkatchenko
Computer Physics Communications, 2019, 240, 38–45.
DOI: 10.1016/j.cpc.2019.02.007
Efficient Training of Machine Learning Potentials by a Randomized Atomic-System Generator
Young-Jae Choi, Seung-Hoon Jhi
The Journal of Physical Chemistry B, 2020, 124 (39), 8704–8710.
DOI: 10/gmf6kr
FCHL Revisited: Faster and More Accurate Quantum Machine Learning
Anders S. Christensen, Lars A. Bratholm, Felix A. Faber, O. Anatole von Lilienfeld
Journal of Chemical Physics, 2020, 152 (4), 044107.
DOI: 10.1063/1.5126701
Gaussian Representation for Image Recognition and Reinforcement Learning of Atomistic Structure
Mads Peter V. Christiansen, Henrik Lund Mortensen, Søren Ager Meldgaard, Bjørk Hammer
Journal of Chemical Physics, 2020, 153 (4).
DOI: 10.1063/5.0015571
Autonomous Discovery in the Chemical Sciences Part I: Progress
Connor W. Coley, Natalie S. Eyke, Klavs F. Jensen
Angewandte Chemie-International Edition, 2020, 59 (51), 22858–22893.
DOI: 10.1002/anie.201909987
Dielectric Response with Short-Ranged Electrostatics
Stephen J. Cox
Proceedings of the National Academy of Sciences, 2020, 117 (33), 19746–19752.
DOI: 10/ghc8bb
Highly Accurate Many-Body Potentials for Simulations of N2O5 in Water: Benchmarks, Development, and Validation
Vinicius Wilian D. Cruzeiro, Eleftherios Lambros, Marc Riera, Ronak Roy, Francesco Paesani, Andreas W. Gotz
Journal of Chemical Theory and Computation, 2021, 17 (7), 3931–3945.
DOI: 10.1021/acs.jctc.1c00069
Analytical Model of Electron Density and Its Machine Learning Inference
Bruno Cuevas-Zuviria, Luis F. Pacios
Journal of Chemical Information and Modeling, 2020, 60 (8), 3831–3842.
DOI: 10.1021/acs.jcim.0c00197
Large Deviations for the Perceptron Model and Consequences for Active Learning
H Cui, L Saglietti, L Zdeborová - Mathematical and Scientific, undefined 2020
proceedings.mlr.press, 2020, 107, 390–430.
Biomolecular QM/MM Simulations: What Are Some of the "Burning Issues"?
Qiang Cui, Tanmoy Pal, Luke Xie
Journal of Physical Chemistry B, 2021, 125 (3), 689–702.
DOI: 10.1021/acs.jpcb.0c09898
Grain Boundary Strengthening in ZrB2 by Segregation of W: Atomistic Simulations with Deep Learning Potential
Fu-Zhi Dai, Bo Wen, Huimin Xiang, Yanchun Zhou
Journal of the European Ceramic Society, 2020, 40 (15), 5029–5036.
DOI: 10.1016/j.jeurceramsoc.2020.06.007
Temperature Dependent Thermal and Elastic Properties of High Entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B-2: Molecular Dynamics Simulation by Deep Learning Potential
Fu-Zhi Dai, Yinjie Sun, Bo Wen, Huimin Xiang, Yanchun Zhou
Journal of Materials Science \& Technology, 2021, 72, 8–15.
DOI: 10.1016/j.jmst.2020.07.014
Theoretical Prediction on Thermal and Mechanical Properties of High Entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by Deep Learning Potential
Fu-Zhi Dai, Bo Wen, Yinjie Sun, Huimin Xiang, Yanchun Zhou
Journal of Materials Science \& Technology, 2020, 43, 168–174.
DOI: 10.1016/j.jmst.2020.01.005
Relationship of Structure and Mechanical Property of Silica with Enhanced Sampling and Machine Learning
Yuanpeng Deng, Tao Du, Hui Li
Journal of the American Ceramic Society, 2021, 104 (8), 3910–3920.
DOI: 10/gmfw49
A General-Purpose Machine-Learning Force Field for Bulk and Nanostructured Phosphorus
Volker L. Deringer, Miguel A. Caro, Gabor Csanyi
Nature Communications, 2020, 11 (1), 5461.
DOI: 10.1038/s41467-020-19168-z
Modelling and Understanding Battery Materials with Machine-Learning-Driven Atomistic Simulations
Volker L. Deringer
Journal of Physics-Energy, 2020, 2 (4), 041003.
DOI: 10.1088/2515-7655/abb011
Learning from the Density to Correct Total Energy and Forces in First Principle Simulations
Sebastian Dick, Marivi Fernandez-Serra
The Journal of Chemical Physics, 2019, 151 (14), 144102.
DOI: 10/gmgftv
Hierarchical Machine Learning of Potential Energy Surfaces
Pavlo O. Dral, Alec Owens, Alexey Dral, Gabor Csanyi
Journal of Chemical Physics, 2020, 152 (20).
DOI: 10.1063/5.0006498
MLatom 2: An Integrative Platform for Atomistic Machine Learning
Pavlo O. Dral, Fuchun Ge, Bao-Xin Xue, Yi-Fan Hou, Max Pinheiro, Jianxing Huang, Mario Barbatti
Topics in Current Chemistry, 2021, 379 (4), 27.
DOI: 10.1007/s41061-021-00339-5
Quantum Chemistry in the Age of Machine Learning
Pavlo O. Dral
Journal of Physical Chemistry Letters, 2020, 11 (6), 2336–2347.
DOI: 10.1021/acs.jpclett.9b03664
Toward Efficient Generation, Correction, and Properties Control of Unique Drug-like Structures
Maksym Druchok, Dzvenymyra Yarish, Oleksandr Gurbych, Mykola Maksymenko
Journal of Computational Chemistry, 2021, 42 (11), 746–760.
DOI: 10.1002/jcc.26494
Dynamics \& Spectroscopy with Neutrons-Recent Developments \& Emerging Opportunities
Kacper Druzbicki, Mattia Gaboardi, Felix Fernandez-Alonso
Polymers, 2021, 13 (9), 1440.
DOI: 10.3390/polym13091440
Data-Driven Approaches Can Overcome the Cost-Accuracy Trade-Off in Multireference Diagnostics
Chenru Duan, Fang Liu, Aditya Nandy, Heather J. Kulik
Journal of Chemical Theory and Computation, 2020, 16 (7), 4373–4387.
DOI: 10.1021/acs.jctc.0c00358
Learning from Failure: Predicting Electronic Structure Calculation Outcomes with Machine Learning Models
Chenru Duan, Jon Paul Janet, Fang Liu, Aditya Nandy, Heather J. Kulik
Journal of Chemical Theory and Computation, 2019, 15 (4), 2331–2345.
DOI: 10.1021/acs.jctc.9b00057
Design, Parameterization, and Implementation of Atomic Force Fields for Adsorption in Nanoporous Materials
D Dubbeldam, KS Walton, TJH Vlugt - Advanced Theory and …, undefined 2019
Wiley Online Library, 2019, 2 (11).
DOI: 10.1002/adts.201900135
Atomic Cluster Expansion: Completeness, Efficiency and Stability
Genevieve Dusson, Markus Bachmayr, Gabor Csanyi, Ralf Drautz, Simon Etter, Cas van der Oord, Christoph Ortner
2021.
Algorithms for Solving High Dimensional PDEs: From Nonlinear Monte Carlo to Machine Learning
Weinan E, Jiequn Han, Arnulf Jentzen, A Jentzen - arXiv preprint ArXiv:2008.13333, undefined 2020
arxiv.org, 2020.
Accelerating Finite-Temperature Kohn-Sham Density Functional Theory with Deep Neural Networks
J. A. Ellis, L. Fiedler, G. A. Popoola, N. A. Modine, J. A. Stephens, A. P. Thompson, A. Cangi, S. Rajamanickam
Physical Review B, 2021, 104 (3), 035120.
DOI: 10.1103/PhysRevB.104.035120
Neuroevolution Machine Learning Potentials: Combining High Accuracy and Low Cost in Atomistic Simulations and Application to Heat Transport
Zheyong Fan, Zezhu Zeng, Cunzhi Zhang, Yanzhou Wang, Haikuan Dong, Yue Chen, Tapio Ala-Nissila
2021.
A Mathematical Principle of Deep Learning: Learn the Geodesic Curve in the Wasserstein Space
Kuo Gai, Shihua Zhang
2021.
Reactive Uptake of N2O5 by Atmospheric Aerosol Is Dominated by Interfacial Processes
M Galib, DT Limmer
science.sciencemag.org, 2021.
Deep Learning in Protein Structural Modeling and Design
Wenhao Gao, Sai Pooja Mahajan, Jeremias Sulam, Jeffrey J. Gray
Patterns, 2020, 1 (9), 100142.
DOI: 10.1016/j.patter.2020.100142
Short Solvent Model for Ion Correlations and Hydrophobic Association
Ang Gao, Richard C. Remsing, John D. Weeks
Proceedings of the National Academy of Sciences of the United States of America, 2020, 117 (3), 1293–1302.
DOI: 10.1073/pnas.1918981117
TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials
Xiang Gao, Farhad Ramezanghorbani, Olexandr Isayev, Justin S. Smith, Adrian E. Roitberg
Journal of Chemical Information and Modeling, 2020, 60 (7), 3408–3415.
DOI: 10.1021/acs.jcim.0c00451
Signatures of a Liquid-Liquid Transition in an Ab Initio Deep Neural Network Model for Water
Thomas E. Gartner, Linfeng Zhang, Pablo M. Piaggi, Roberto Car, Athanassios Z. Panagiotopoulos, Pablo G. Debenedetti
Proceedings of the National Academy of Sciences of the United States of America, 2020, 117 (42), 26040–26046.
DOI: 10.1073/pnas.2015440117
Combining Phonon Accuracy with High Transferability in Gaussian Approximation Potential Models
Janine George, Geoffroy Hautier, Albert P. Bartok, Gabor Csanyi, Volker L. Deringer
Journal of Chemical Physics, 2020, 153 (4), 044104.
DOI: 10.1063/5.0013826
The Role of Feature Space in Atomistic Learning
Alexander Goscinski, Guillaume Fraux, Giulio Imbalzano, Michele Ceriotti
Machine Learning-Science and Technology, 2021, 2 (2), 025028.
DOI: 10.1088/2632-2153/abdaf7
Code Interoperability Extends the Scope of Quantum Simulations
Marco Govoni, Jonathan Whitmer, Juan de Pablo, Francois Gygi, Giulia Galli
Npj Computational Materials, 2021, 7 (1), 32.
DOI: 10.1038/s41524-021-00501-z
Incorporating Long-Range Physics in Atomic-Scale Machine Learning
Andrea Grisafi, Michele Ceriotti
Journal of Chemical Physics, 2019, 151 (20), 204105.
DOI: 10.1063/1.5128375
Multi-Scale Approach for the Prediction of Atomic Scale Properties
Andrea Grisafi, Jigyasa Nigam, Michele Ceriotti
Chemical Science, 2021, 12 (6), 2078–2090.
DOI: 10.1039/d0sc04934d
Deep Neural Network Model for Approximating Eigenmodes Localized by a Confining Potential
L Grubišić, M Hajba, D Lacmanović - Entropy
mdpi.com, 2021, 2, 27001.
DOI: 10.1088/2632-2153/abc940
Finite-Temperature Interplay of Structural Stability, Chemical Complexity, and Elastic Properties of Bcc Multicomponent Alloys from Ab Initio Trained Machine-Learning Potentials
Konstantin Gubaev, Yuji Ikeda, Ferenc Tasnadi, Joerg Neugebauer, Alexander Shapeev, Blazej Grabowski, Fritz Koermann
Physical Review Materials, 2021, 5 (7), 073801.
DOI: 10.1103/PhysRevMaterials.5.073801
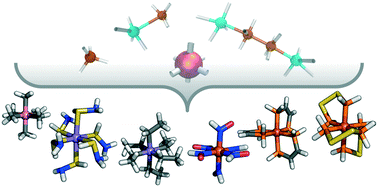
Enumeration of de Novo Inorganic Complexes for Chemical Discovery and Machine Learning
Stefan Gugler, Jon Paul Janet, Heather J. Kulik
Molecular Systems Design \& Engineering, 2020, 5 (1), 139–152.
DOI: 10.1039/c9me00069k
High-Repetition-Rate Femtosecond Mid-Infrared Pulses Generated by Nonlinear Optical Modulation of Continuous-Wave QCLs and ICLs
Chenglin Gu, Zhong Zuo, Daping Luo, Daowang Peng, Yuanfeng Di, Xing Zou, Liu Yang, Wenxue Li
Optics Letters, 2019, 44 (23), 5848–5851.
DOI: 10.1364/OL.44.005848
Neural Network Representation of Electronic Structure from Ab Initio Molecular Dynamics
Q Gu, L Zhang, J Feng
arxiv.org, 2021.
Bergman-Type Medium Range Order in Amorphous Zr77Rh23 Alloy Studied by Ab Initio Molecular Dynamics Simulations
Y. R. Guo, Chong Qiao, J. J. Wang, H. Shen, S. Y. Wang, Y. X. Zheng, R. J. Zhang, L. Y. Chen, Wan-Sheng Su, C. Z. Wang, K. M. Ho
Journal of Alloys and Compounds, 2019, 790, 675–682.
DOI: 10.1016/j.jallcom.2019.03.197
The Thermoelectric Performance of New Structure SnSe Studied by Quotient Graph and Deep Learning Potential
D. Guo, C. Li, K. Li, B. Shao, D. Chen, Y. Ma, J. Sun, X. Cao, W. Zeng, X. Chang
Materials Today Energy, 2021, 20, 100665.
DOI: 10/gmgd38
Sparse Gaussian Process Potentials: Application to Lithium Diffusivity in Superionic Conducting Solid Electrolytes
Amir Hajibabaei, Chang Woo Myung, Kwang S. Kim
Physical Review B, 2021, 103 (21), 214102.
DOI: 10.1103/PhysRevB.103.214102
MAISE: Construction of Neural Network Interatomic Models and Evolutionary Structure Optimization
S Hajinazar, A Thorn, ED Sandoval
Elsevier, 2020.
Machine Learning-Assisted Excited State Molecular Dynamics with the State-Interaction State-Averaged Spin-Restricted Ensemble-Referenced Kohn-Sham Approach
Jong-Kwon Ha, Kicheol Kim, Seung Kyu Min
Journal of Chemical Theory and Computation, 2021, 17 (2), 694–702.
DOI: 10.1021/acs.jctc.0c01261
Dynamic Observation of Dendritic Quasicrystal Growth upon Laser-Induced Solid-State Transformation
Insung Han, Joseph T. McKeown, Ling Tang, Cai-Zhuang Wang, Hadi Parsamehr, Zhucong Xi, Ying-Rui Lu, Matthew J. Kramer, Ashwin J. Shahani
Physical Review Letters, 2020, 125 (19), 195503.
DOI: 10.1103/PhysRevLett.125.195503
A Machine Learning Approach for MP2 Correlation Energies and Its Application to Organic Compounds
Ruocheng Han, Mauricio Rodriguez-Mayorga, Sandra Luber
Journal of Chemical Theory and Computation, 2021, 17 (2), 777–790.
DOI: 10.1021/acs.jctc.0c00898
Solving Many-Electron Schrodinger Equation Using Deep Neural Networks
Jiequn Han, Linfeng Zhang, Weinan E
Journal of Computational Physics, 2019, 399, 108929.
DOI: 10.1016/j.jcp.2019.108929
Trajectory-Based Machine Learning Method and Its Application to Molecular Dynamics
R. Han, S. Luber
Molecular Physics, 2020, 118 (19-20).
DOI: 10.1080/00268976.2020.1788189
Uniformly Accurate Machine Learning-Based Hydrodynamic Models for Kinetic Equations
Jiequn Han, Chao Ma, Zheng Ma, Weinan E
Proceedings of the National Academy of Sciences of the United States of America, 2019, 116 (44), 21983–21991.
DOI: 10.1073/pnas.1909854116
Uniformly Accurate Machine Learning-Based Hydrodynamic Models for Kinetic Equations
Jiequn Han, Chao Ma, Zheng Ma, Weinan E
Proceedings of the National Academy of Sciences of the United States of America, 2019, 116 (44), 21983–21991.
DOI: 10.1073/pnas.1909854116
Universal Approximation of Symmetric and Anti-Symmetric Functions
J Han, Y Li, L Lin, J Lu, J Zhang, L Zhang
arxiv.org, 2019.
Validating First-Principles Molecular Dynamics Calculations of Oxide/Water Interfaces with x-Ray Reflectivity Data
Katherine J. Harmon, Kendra Letchworth-Weaver, Alex P. Gaiduk, Federico Giberti, Francois Gygi, Maria K. Y. Chan, Paul Fenter, Giulia Galli
Physical Review Materials, 2020, 4 (11), 113805.
DOI: 10.1103/PhysRevMaterials.4.113805
An Open Combinatorial Diffraction Dataset Including Consensus Human and Machine Learning Labels with Quantified Uncertainty for Training New Machine Learning Models
Jason R. Hattrick-Simpers, Brian DeCost, A. Gilad Kusne, Howie Joress, Winnie Wong-Ng, Debra L. Kaiser, Andriy Zakutayev, Caleb Phillips, Shijing Sun, Janak Thapa, Heshan Yu, Ichiro Takeuchi, Tonio Buonassisi
Integrating Materials and Manufacturing Innovation, 2021, 10 (2), 311–318.
DOI: 10/gkhbw2

Fast, Accurate, and Transferable Many-Body Interatomic Potentials by Symbolic Regression
Alberto Hernandez, Adarsh Balasubramanian, Fenglin Yuan, Simon A. M. Mason, Tim Mueller
Npj Computational Materials, 2019, 5, 112.
DOI: 10.1038/s41524-019-0249-1
Compressing Physical Properties of Atomic Species for Improving Predictive Chemistry
John E. Herr, Kevin Koh, Kun Yao, John Parkhill
The Journal of Chemical Physics, 2019, 151 (8), 084103.
DOI: 10/ggb5bq
Compressing Physics with an Autoencoder: Creating an Atomic Species Representation to Improve Machine Learning Models in the Chemical Sciences
John E. Herr, Kevin Koh, Kun Yao, John Parkhill
Journal of Chemical Physics, 2019, 151 (8), 084103.
DOI: 10.1063/1.5108803
In Operando Active Learning of Interatomic Interaction during Large-Scale Simulations
M Hodapp, A Shapeev - Machine Learning: Science And, undefined 2020
iopscience.iop.org, 2020.
DOI: 10.1088/2632-2153/aba373
Machine-Learning Potentials Enable Predictive \$\textbackslash textit{and}\$ Tractable High-Throughput Screening of Random Alloys
Max Hodapp, Alexander Shapeev
2021.
Dielectric Constant of Supercritical Water in a Large Pressure-Temperature Range
Rui Hou, Yuhui Quan, Ding Pan
Journal of Chemical Physics, 2020, 153 (10), 101103.
DOI: 10.1063/5.0020811
Deep Potential Generation Scheme and Simulation Protocol for the Li10GeP2S12-Type Superionic Conductors
Jianxing Huang, Linfeng Zhang, Han Wang, Jinbao Zhao, Jun Cheng, E. Weinan
Journal of Chemical Physics, 2021, 154 (9), 094703.
DOI: 10.1063/5.0041849
Ab Initio Machine Learning in Chemical Compound Space
Bing Huang, O. Anatole von Lilienfeld
2021.
Int-Deep: A Deep Learning Initialized Iterative Method for Nonlinear Problems
Jianguo Huang, Haoqin Wang, Haizhao Yang
Journal of Computational Physics, 2020, 419, 109675.
DOI: 10/gg2rtj
Learning Thermodynamically Stable and Galilean Invariant Partial Differential Equations for Non-Equilibrium Flows
Juntao Huang, Zhiting Ma, Yizhou Zhou, Wen An Yong
Journal of Non-Equilibrium Thermodynamics, 2021.
DOI: 10.1515/JNET-2021-0008/HTML
Machine Learning Moment Closure Models for the Radiative Transfer Equation I: Directly Learning a Gradient Based Closure
Juntao Huang, Yingda Cheng, Andrew J. Christlieb, Luke F. Roberts
2021.
Inclusion of Machine Learning Kernel Ridge Regression Potential Energy Surfaces in On-the-Fly Nonadiabatic Molecular Dynamics Simulation
Deping Hu, Yu Xie, Xusong Li, Lingyue Li, Zhenggang Lan
Journal of Physical Chemistry Letters, 2018, 9 (11), 2725–2732.
DOI: 10.1021/acs.jpclett.8b00684
Neural Network Force Fields for Metal Growth Based on Energy Decompositions
Qin Hu, Mouyi Weng, Xin Chen, Shucheng Li, Feng Pan, Lin-Wang Wang
Journal of Physical Chemistry Letters, 2020, 11 (4), 364–1369.
DOI: 10.1021/acs.jpclett.9b03780
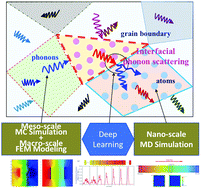
Perspective on Multi-Scale Simulation of Thermal Transport in Solids and Interfaces
Ming Hu, Zhonghua Yang
Physical Chemistry Chemical Physics, 2021, 23 (3), 1785–1801.
DOI: 10.1039/d0cp03372c
Coarse Graining Molecular Dynamics with Graph Neural Networks
Brooke E. Husic, Nicholas E. Charron, Dominik Lemm, Jiang Wang, Adria Perez, Maciej Majewski, Andreas Kramer, Yaoyi Chen, Simon Olsson, Gianni de Fabritiis, Frank Noe, Cecilia Clementi
Journal of Chemical Physics, 2020, 153 (19), 194101.
DOI: 10.1063/5.0026133
Artificial Neutral Networks (ANNs) Applied as CFD Optimization Techniques
Ideen Sadrehaghighi
2021.
DOI: 10/gmf5vh
Efficient Multiscale Optoelectronic Prediction for Conjugated Polymers
Nicholas E. Jackson, Alec S. Bowen, Juan J. de Pablo
Macromolecules, 2020, 53 (1), 482–490.
DOI: 10.1021/acs.macromol.9b02020
Electronic Structure at Coarse-Grained Resolutions from Supervised Machine Learning
Nicholas E. Jackson, Alec S. Bowen, Lucas W. Antony, Michael A. Webb, Venkatram Vishwanath, Juan J. de Pablo
Science Advances, 2019, 5 (3), eaav1190.
DOI: 10.1126/sciadv.aav1190
Recent Advances in Machine Learning towards Multiscale Soft Materials Design
Nicholas E. Jackson, Michael A. Webb, Juan J. de Pablo
Current Opinion in Chemical Engineering, 2019, 23, 106–114.
DOI: 10.1016/j.coche.2019.03.005
Machine Learning for Metallurgy III: A Neural Network Potential for Al-Mg-Si
Abhinav C.P. Jain, Daniel Marchand, Albert Glensk, M. Ceriotti, W. A. Curtin
Physical Review Materials, 2021, 5 (5).
DOI: 10.1103/physrevmaterials.5.053805
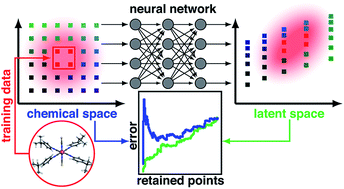
A Quantitative Uncertainty Metric Controls Error in Neural Network-Driven Chemical Discovery
Jon Paul Janet, Chenru Duan, Tzuhsiung Yang, Aditya Nandy, Heather J. Kulik
Chemical Science, 2019, 10 (34), 7913–7922.
DOI: 10.1039/c9sc02298h
Uncertain Times Call for Quantitative Uncertainty Metrics: Controlling Error in Neural Network Predictions for Chemical Discovery
Jon Paul Janet, Chenru Duan, Tzuhsiung Yang, Aditya Nandy, Heather Kulik
2019.
DOI: 10.26434/chemrxiv.7900277.v1
Towards Fully Ab Initio Simulation of Atmospheric Aerosol Nucleation
S Jiang, YR Liu, T Huang, YJ Feng, CY Wang
arxiv.org, 2021.
Accurate Deep Potential Model for the Al–Cu–Mg Alloy in the Full Concentration Space
W Jiang, Y Zhang, L Zhang, Wang H
iopscience.iop.org, 2021.
Accurate Deep Potential Model for the Al-Cu-Mg Alloy in the Full Concentration Space
Wanrun Jiang, Yuzhi Zhang, Linfeng Zhang, Han Wang
Chinese Physics B, 2021, 30 (5), 050706.
DOI: 10.1088/1674-1056/abf134
Self-Healing Mechanism of Lithium Metal
Junyu Jiao, Genming Lai, Jiaze Lu, Xianqi Xu, Jing Wang, Jiaxin Zheng
2021.
Pushing the Limit of Molecular Dynamics with Ab Initio Accuracy to 100 Million Atoms with Machine Learning
Weile Jia, Han Wang, Mohan Chen, Denghui Lu, L Lin, Lin Lin, Roberto Car, Linfeng Zhang
ieeexplore.ieee.org, 2021.
On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations
Ryosuke Jinnouchi, Kazutoshi Miwa, Ferenc Karsai, Georg Kresse, Ryoji Asahi
Journal of Physical Chemistry Letters, 2020, 11 (17), 6946–6955.
DOI: 10.1021/acs.jpclett.0c01061
Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration
Pei-Lin Kang, Cheng Shang, Zhi-Pan Liu
Accounts of Chemical Research, 2020, 53 (10), 2119–2129.
DOI: 10.1021/acs.accounts.0c00472
Enabling Ab Initio Configurational Sampling of Multicomponent Solids with Long-Range Interactions Using Neural Network Potentials and Active Learning
Shusuke Kasamatsu, Yuichi Motoyama, Kazuyoshi Yoshimi, Ushio Matsumoto, Akihide Kuwabara, Takafumi Ogawa
2020.
Combining Machine Learning and Computational Chemistry for Predictive Insights Into Chemical Systems
John A. Keith, Valentin Vassilev-Galindo, Bingqing Cheng, Stefan Chmiela, Michael Gastegger, Klaus-Robert Müller, Alexandre Tkatchenko
2021.
Reaction Path-Force Matching in Collective Variables: Determining Ab Initio QM/MM Free Energy Profiles by Fitting Mean Force
Bryant Kim, Ryan Snyder, Mulpuri Nagaraju, Yan Zhou, Pedro Ojeda-May, Seth Keeton, Mellisa Hege, Yihan Shao, Jingzhi Pu
Journal of Chemical Theory and Computation, 2021, 17 (8), 4961–4980.
DOI: 10/gmfw5p
Neural Network Potentials: A Concise Overview of Methods
Emir Kocer, TW Tsz Wai Ko, Jörg Behler, J Behler
arxiv.org, 2021.
Enabling Large-Scale Condensed-Phase Hybrid Density Functional Theory Based Ab Initio Molecular Dynamics. 1. Theory, Algorithm, and Performance
Hsin-Yu Ko, Junteng Jia, Biswajit Santra, Xifan Wu, Roberto Car, Robert DiStasio
Journal of Chemical Theory and Computation, 2020, 16 (6), 3757–3785.
DOI: 10.1021/acs.jctc.9b01167
Isotope Effects in Liquid Water via Deep Potential Molecular Dynamics
Hsin-Yu Ko, Linfeng Zhang, Biswajit Santra, Han Wang, Weinan E, Robert A. DiStasio, Roberto Car
Molecular Physics, 2019, 117 (22), 3269–3281.
DOI: 10.1080/00268976.2019.1652366
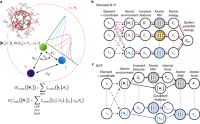
N-Body Networks: A Covariant Hierarchical Neural Network Architecture for Learning Atomic Potentials
Risi Kondor
2018.
Manifold Learning for Coarse-Graining Atomistic Simulations: Application to Amorphous Solids
Katiana Kontolati, Darius Alix-Williams, Nicholas M. Boffi, Michael L. Falk, Chris H. Rycroft, Michael D. Shields
2021.
Accessing Thermal Conductivity of Complex Compounds by Machine Learning Interatomic Potentials
P Korotaev, I Novoselov, A Yanilkin, A Shapeev B
APS, 2019, 100 (14), 144308.
DOI: 10.1103/physrevb.100.144308
Dielectric Constant of Liquid Water Determined with Neural Network Quantum Molecular Dynamics
Aravind Krishnamoorthy, Ken-ichi Nomura, Nitish Baradwaj, Kohei Shimamura, Pankaj Rajak, Ankit Mishra, Shogo Fukushima, Fuyuki Shimojo, Rajiv Kalia, Aiichiro Nakano, Priya Vashishta
Physical Review Letters, 2021, 126 (21), 216403.
DOI: 10.1103/PhysRevLett.126.216403
Size and Temperature Transferability of Direct and Local Deep Neural Networks for Atomic Forces
Natalia Kuritz, Goren Gordon, Amir Natan
Physical Review B, 2018, 98 (9), 094109.
DOI: 10/gkv2j9
The Estimation of the Second Virial Coefficients of He and N2 Based on Neural Network Potentials with Quantum Mechanical Calculations
Taejin Kwon, Han Wook Song, Sam Yong Woo, Jong-Ho Kim, Bong June Sung
Chemical Physics, 2021, 548, 111231.
DOI: 10/gmf6ws
Machine-Learning-Based Non-Newtonian Fluid Model with Molecular Fidelity
Huan Lei, Lei Wu, Weinan Weinan
Physical Review E, 2020, 102 (4).
DOI: 10.1103/physreve.102.043309
Modeling Electrochemical Interfaces from Ab Initio Molecular Dynamics: Water Adsorption on Metal Surfaces at Potential of Zero Charge
Jia-Bo Le, Jun Cheng
Current Opinion in Electrochemistry, 2020, 19, 129–136.
DOI: 10.1016/j.coelec.2019.11.008
Non-Classical Nucleation Pathways in Stacking-Disordered Crystals
Fabio Leoni, John Russo
2021.
Nonclassical Nucleation Pathways in Stacking-Disordered Crystals
Fabio Leoni, John Russo
Physical Review X, 2021, 11 (3), 031006.
DOI: 10.1103/PhysRevX.11.031006
Accurate and Transferable Reactive Molecular Dynamics Models from Constrained Density Functional Theory
Chenghan Li, Gregory A Voth
, 31.
Analysis of Trajectory Similarity and Configuration Similarity in On-the-Fly Surface-Hopping Simulation on Multi-Channel Nonadiabatic Photoisomerization Dynamics
Xusong Li, Deping Hu, Yu Xie, Zhenggang Lan
Journal of Chemical Physics, 2018, 149 (24), 244104.
DOI: 10.1063/1.5048049
Machine-Learning-Driven Simulations on Microstructure and Thermophysical Properties of MgCl2-KCl Eutectic
Wenshuo Liang, Guimin Lu, Jianguo Yu
Acs Applied Materials \& Interfaces, 2021, 13 (3), 4034–4042.
DOI: 10.1021/acsami.0c20665
Molecular Dynamics Simulations of Molten Magnesium Chloride Using Machine-Learning-Based Deep Potential
Wenshuo Liang, Guimin Lu, Jianguo Yu
Advanced Theory and Simulations, 2020, 3 (12), 2000180.
DOI: 10.1002/adts.202000180
Theoretical Prediction on the Local Structure and Transport Properties of Molten Alkali Chlorides by Deep Potentials
Wenshuo Liang, Guimin Lu, Jianguo Yu
Journal of Materials Science \& Technology, 2021, 75, 78–85.
DOI: 10/gmf63v
Better Approximations of High Dimensional Smooth Functions by Deep Neural Networks with Rectified Power Units
Bo Li, Shanshan Tang, Haijun Yu
Communications in Computational Physics, 2020, 27 (2), 379–411.
DOI: 10.4208/cicp.OA-2019-0168
CONFORMATION-GUIDED MOLECULAR REPRESENTA- TION WITH HAMILTONIAN NEURAL NETWORKS
Ziyao Li, Shuwen Yang, Guojie Song, Lingsheng Cai
2021, 11.
Development of Robust Neural-Network Interatomic Potential for Molten Salt
Qing-Jie Li, Emine Kucukbenli, Stephen Lam, Boris Khaykovich, Efthimios Kaxiras, Ju Li
Cell Reports Physical Science, 2021, 2 (3), 100359.
DOI: 10.1016/j.xcrp.2021.100359
Effect of Local Structural Disorder on Lithium Diffusion Behavior in Amorphous Silicon
Wenwen Li, Yasunobu Ando
Physical Review Materials, 2020, 4 (4).
DOI: 10.1103/physrevmaterials.4.045602
HamNet: Conformation-Guided Molecular Representation with Hamiltonian Neural Networks
Ziyao Li, Shuwen Yang, Guojie Song, Lingsheng Cai
2021.
Introducing Block Design in Graph Neural Networks for Molecular Properties Prediction
Yuquan Li, Pengyong Li, Xing Yang, Chang-Yu Hsieh, Shengyu Zhang, Xiaorui Wang, Ruiqiang Lu, Huanxiang Liu, Xiaojun Yao
Chemical Engineering Journal, 2021, 414, 128817.
DOI: 10.1016/j.cej.2021.128817
Machine Learning in Computational Surface Science and Catalysis: Case Studies on Water and Metal–Oxide Interfaces
Xiaoke Li, Wolfgang Paier, Joachim Paier
Frontiers in Chemistry, 2020, 8, 601029.
DOI: 10/ghnggc
Multilevel Fine-Tuning: Closing Generalization Gaps in Approximation of Solution Maps Under a Limited Budget for Training Data
Zhihan Li, Yuwei Fan, Lexing Ying
Multiscale Modeling \& Simulation, 2021, 19 (1), 344–373.
DOI: 10.1137/20M1326404
Neural Canonical Transformation with Symplectic Flows
Shuo-Hui Li, Chen-Xiao Dong, Linfeng Zhang, Lei Wang
Physical Review X, 2020, 10 (2), 021020.
DOI: 10.1103/PhysRevX.10.021020
A Neural-Network Based Framework of Developing Cross Interaction in Alloy Embedded-Atom Method Potentials: Application to Zr-Nb Alloy
Bo Lin, Jincheng Wang, Junjie Li, Zhijun Wang
Journal of Physics-Condensed Matter, 2021, 33 (8), 084004.
DOI: 10.1088/1361-648X/abcb69
Numerical Methods for Kohn-Sham Density Functional Theory
Lin Lin, Jianfeng Lu, Lexing Ying
Acta Numerica, 2019, 28, 405–539.
DOI: 10.1017/S0962492919000047
Searching Configurations in Uncertainty Space: Active Learning of High-Dimensional Neural Network Reactive Potentials
Qidong Lin, Liang Zhang, Yaolong Zhang, Bin Jiang
Journal of Chemical Theory and Computation, 2021, 17 (5), 2691–2701.
DOI: 10/gmfw5n
Unravelling the Fast Alkali-Ion Dynamics in Paramagnetic Battery Materials Combined with NMR and Deep-Potential Molecular Dynamics Simulation
Min Lin, Xiangsi Liu, Yuxuan Xiang, Feng Wang, Yunpei Liu, Riqiang Fu, Jun Cheng, Yong Yang
Angewandte Chemie-International Edition, 2021, 60 (22), 12547–12553.
DOI: 10.1002/anie.202102740
PowerNet: Efficient Representations of Polynomials and Smooth Functions by Deep Neural Networks with Rectified Power Units
Bo Li, Shanshan Tang, Haijun Yu
Journal of Mathematical Study, 2020, 53 (2), 159–191.
DOI: 10.4208/jms.v53n2.20.03
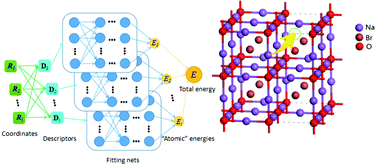
Theoretical Study of Na+ Transport in the Solid-State Electrolyte Na3OBr Based on Deep Potential Molecular Dynamics
Han-Xiao Li, Xu-Yuan Zhou, Yue-Chao Wang, Hong Jiang
Inorganic Chemistry Frontiers, 2021, 8 (2), 425–432.
DOI: 10.1039/d0qi00921k
Machine Learning Phase Space Quantum Dynamics Approaches
Xinzijian Liu, Linfeng Zhang, Jian Liu
Journal of Chemical Physics, 2021, 154 (18), 184104.
DOI: 10.1063/5.0046689
A Unified Deep Neural Network Potential Capable of Predicting Thermal Conductivity of Silicon in Different Phases
R. Li, E. Lee, T. Luo
Materials Today Physics, 2020, 12, 100181.
DOI: 10.1016/j.mtphys.2020.100181
Rapid Detection of Strong Correlation with Machine Learning for Transition-Metal Complex High-Throughput Screening
Fang Liu, Chenru Duan, Heather J. Kulik
Journal of Physical Chemistry Letters, 2020, 11 (19), 8067–8076.
DOI: 10.1021/acs.jpclett.0c02288
Structure and Dynamics of Warm Dense Aluminum: A Molecular Dynamics Study with Density Functional Theory and Deep Potential
Qianrui Liu, Denghui Lu, Mohan Chen
Journal of Physics-Condensed Matter, 2020, 32 (14), 144002.
DOI: 10.1088/1361-648X/ab5890
Thermal Transport by Electrons and Ions in Warm Dense Aluminum: A Combined Density Functional Theory and Deep Potential Study
Qianrui Liu, Junyi Li, Mohan Chen
Matter and Radiation at Extremes, 2021, 6 (2).
DOI: 10.1063/5.0030123
Transferable Multilevel Attention Neural Network for Accurate Prediction of Quantum Chemistry Properties via Multitask Learning
Ziteng Liu, Liqiang Lin, Qingqing Jia, Zheng Cheng, Yanyan Jiang, Yanwen Guo, Jing Ma
Journal of Chemical Information and Modeling, 2021, 61 (3), 1066–1082.
DOI: 10.1021/acs.jcim.0c01224
Active Learning a Coarse-Grained Neural Network Model for Bulk Water from Sparse Training Data
TD Loeffler, TK Patra, Chan H
pubs.rsc.org.
Active Learning a Neural Network Model for Gold Clusters\& Bulk from Sparse First Principles Training Data
TD Loeffler, S Manna, TK Patra, Chan H
arxiv.org, 2020.
Active Learning the Potential Energy Landscape for Water Clusters from Sparse Training Data
Troy D. Loeffler, Tarak K. Patra, Henry Chan, Mathew Cherukara, Subramanian K.R.S. Sankaranarayanan
Journal of Physical Chemistry C, 2020, 124 (8), 4907–4916.
DOI: 10.1021/acs.jpcc.0c00047
PDE-Net 2.0: Learning PDEs from Data with a Numeric-Symbolic Hybrid Deep Network
Zichao Long, Yiping Lu, Bin Dong
Journal of Computational Physics, 2019, 399, 108925.
DOI: 10.1016/j.jcp.2019.108925
PANNA: Properties from Artificial Neural Network Architectures
Ruggero Lot, Franco Pellegrini, Yusuf Shaidu, Emine Kucukbenli
Computer Physics Communications, 2020, 256, 107402.
DOI: 10.1016/j.cpc.2020.107402
Deep Learning: New Engine for the Study of Material Microstructures and Physical Properties
G Lu, S Duan
Modern Physics 现代物理, 2019, 2019 (6), 263–276.
DOI: 10.12677/mp.2019.96026
Dataset Construction to Explore Chemical Space with 3D Geometry and Deep Learning
Jianing Lu, Song Xia, Jieyu Lu, Yingkai Zhang
Journal of Chemical Information and Modeling, 2021, 61 (3), 1095–1104.
DOI: 10.1021/acs.jcim.1c00007
Deep Potential Molecular Dynamics Simulation of 100 Million Atoms with Ab Initio Accuracy
Denghui Lu, Han Wang, Mohan Chen, Lin Lin, Roberto Car, Weinan E, Weile Jia, Linfeng Zhang
Computer Physics Communications, 2021, 259, 107624.
DOI: 10.1016/j.cpc.2020.107624
Deep Potential Molecular Dynamics Simulation of 100 Million Atoms with Ab Initio Accuracy
Denghui Lu, Han Wang, Mohan Chen, Lin Lin, Roberto Car, Weinan E, Weile Jia, Linfeng Zhang
Computer Physics Communications, 2021, 259, 107624.
DOI: 10.1016/j.cpc.2020.107624
DP Train, Then DP Compress: Model Compression in Deep Potential Molecular Dynamics
D Lu, W Jiang, Y Chen, L Zhang, W Jia, H Wang
arxiv.org, 2021.
A Unified Picture of the Covalent Bond within Quantum-Accurate Force Fields: From Organic Molecules to Metallic Complexes' Reactivity
Alessandro Lunghi, Stefano Sanvito
Science Advances, 2019, 5 (5), eaaw2210.
DOI: 10.1126/sciadv.aaw2210
Anomalous Behavior of Viscosity and Electrical Conductivity of MgSiO3 Melt at Mantle Conditions
Haiyang Luo, Bijaya B. Karki, Dipta B. Ghosh, Huiming Bao
Geophysical Research Letters, 2021, 48 (13), e2021GL093573.
DOI: 10/gkrt5v
Deep Neural Network Potentials for Diffusional Lithium Isotope Fractionation in Silicate Melts
Haiyang Luo, Bijaya B. Karki, Dipta B. Ghosh, Huiming Bao
Geochimica et Cosmochimica Acta, 2021, 303, 38–50.
DOI: 10/gmf625
Predicting Molecular Energy Using Force-Field Optimized Geometries and Atomic Vector Representations Learned from an Improved Deep Tensor Neural Network
Jianing Lu, Cheng Wang, Yingkai Zhang
Journal of Chemical Theory and Computation, 2019, 15 (7), 4113–4121.
DOI: 10.1021/acs.jctc.9b00001
Deep Learning Observables in Computational Fluid Dynamics
KO Lye, S Mishra, D Ray - Journal of Computational Physics, undefined 2020
Elsevier, 2019.
A Fast Neural Network Approach for Direct Covariant Forces Prediction in Complex Multi-Element Extended Systems
Jonathan P. Mailoa, Mordechai Kornbluth, Simon Batzner, Georgy Samsonidze, Stephen T. Lam, Jonathan Vandermause, Chris Ablitt, Nicola Molinari, Boris Kozinsky
Nature Machine Intelligence, 2019, 1 (10), 471–479.
DOI: 10.1038/s42256-019-0098-0
Evaluation of Experimental Alkali Metal Ion-Ligand Noncovalent Bond Strengths with DLPNO-CCSD(T) Method
Bholanath Maity, Yury Minenkov, Luigi Cavallo
Journal of Chemical Physics, 2019, 151 (1), 014301.
DOI: 10.1063/1.5099580
Transferability of Neural Network Potentials for Varying Stoichiometry: Phonons and Thermal Conductivity of Mn\$_x\$Ge\$_y\$ Compounds
Claudia Mangold, Shunda Chen, Giuseppe Barbalinardo, Joerg Behler, Pascal Pochet, Konstantinos Termentzidis, Yang Han, Laurent Chaput, David Lacroix, Davide Donadio
Journal of Applied Physics, 2020, 127 (24), 244901.
DOI: 10/gg7jww
Machine Learning for Metallurgy I. A Neural-Network Potential for Al-Cu
Daniel Marchand, Abhinav Jain, Albert Glensk, W. A. Curtin
Physical Review Materials, 2020, 4 (10).
DOI: 10.1103/physrevmaterials.4.103601
Simulating Diffusion Properties of Solid-State Electrolytes via a Neural Network Potential: Performance and Training Scheme
Aris Marcolongo, Tobias Binninger, Federico Zipoli, Teodoro Laino
2019.
Connection between Liquid and Non-Crystalline Solid Phases in Water
Fausto Martelli, Fabio Leoni, Francesco Sciortino, John Russo
Journal of Chemical Physics, 2020, 153 (10), 104503.
DOI: 10.1063/5.0018923
Deep Learning in Chemistry
Adam C. Mater, Michelle L. Coote
Journal of Chemical Information and Modeling, 2019, 59 (6), 2545–2559.
DOI: 10.1021/acs.jcim.9b00266
Machine-Learning Interatomic Potentials for Materials Science
Y Mishin - Acta Materialia, undefined 2021
Elsevier, 2021.
Machine Learning Enhanced Global Optimization by Clustering Local Environments to Enable Bundled Atomic Energies
Soren A. Meldgaard, Esben L. Kolsbjerg, Bjork Hammer
Journal of Chemical Physics, 2018, 149 (13), 134104.
DOI: 10.1063/1.5048290
Transformative Applications of Machine Learning for Chemical Reactions
M. Meuwly
2021.
Liquid to Crystal Si Growth Simulation Using Machine Learning Force Field
Ling Miao, Lin Wang Wang
Journal of Chemical Physics, 2020, 153 (7).
DOI: 10.1063/5.0011163
Strategies for the Construction of Machine-Learning Potentials for Accurate and Efficient Atomic-Scale Simulations
April M. Miksch, Tobias Morawietz, Johannes Kaestner, Alexander Urban, Nongnuch Artrith
Machine Learning-Science and Technology, 2021, 2 (3), 031001.
DOI: 10.1088/2632-2153/abfd96
Gas Phase Silver Thermochemistry from First Principles
Irina Minenkova, Valery V. Slizney, Luigi Cavallo, Yury Minenkov
Inorganic Chemistry, 2019, 58 (12), 7873–7885.
DOI: 10.1021/acs.inorgchem.9b00556
An Automated Approach for Developing Neural Network Interatomic Potentials with FLAME
H Mirhosseini, H Tahmasbi, SR Kuchana - Computational Materials …, undefined 2021
Elsevier, 2021.
Molecular Dynamics Properties without the Full Trajectory: A Denoising Autoencoder Network for Properties of Simple Liquids
Alireza Moradzadeh, N. R. Aluru
Journal of Physical Chemistry Letters, 2019, 10 (24), 7568–7576.
DOI: 10.1021/acs.jpclett.9b02820
Deep Learning for Gravitational-Wave Data Analysis: A Resampling White-Box Approach
Manuel D. Morales, Javier M. Antelis, Claudia Moreno, Alexander I. Nesterov
Sensors, 2021, 21 (9), 3174.
DOI: 10/gmgfm6
Machine Learning-Accelerated Quantum Mechanics-Based Atomistic Simulations for Industrial Applications
Tobias Morawietz, Nongnuch Artrith
Journal of Computer-Aided Molecular Design, 2021, 35 (4), 557–586.
DOI: 10.1007/s10822-020-00346-6
Transfer Learning of Potential Energy Surfaces for Efficient Atomistic Modeling of Doping and Alloy
Pinghui Mo, Mengchao Shi, Wenze Yao, Jie Liu
IEEE Electron Device Letters, 2020, 41 (4), 633–636.
DOI: 10/gg2bfc
Assessment of Localized and Randomized Algorithms for Electronic Structure
Jonathan E. Moussa, Andrew D. Baczewski
Electronic Structure, 2019, 1 (3), 033001.
DOI: 10.1088/2516-1075/ab2022
The Dynamic Control of the Light Signalling Device in Real-Time
Jan Mrazek, Lucia Duricova Mrazkova, Martin Hromada, Jana Reznickova
MATEC Web of Conferences, 2019, 292, 03014.
DOI: 10/gmgfts
Traffic Control Through Traffic Density
Jan Mrazek, Lucia Duricova Mrazkova, Martin Hromada
2019 3rd European Conference on Electrical Engineering and Computer Science (Eecs 2019), 2019, 19–21.
DOI: 10.1109/EECS49779.2019.00017
Machine Learning for Interatomic Potential Models
Tim Mueller, Alberto Hernandez, Chuhong Wang
Journal of Chemical Physics, 2020, 152 (5), 050902.
DOI: 10.1063/1.5126336
Supervised Learning of Few Dirty Bosons with Variable Particle Number
P Mujal, À Martínez Miguel, A Polls
scipost.org, 2020.
Machine Learning at the Atomic Scale
Felix Musil, Michele Ceriotti
Chimia, 2019, 73 (12), 972–982.
DOI: 10.2533/chimia.2019.972
Non-Empirical Weighted Langevin Mechanics for the Potential Escape Problem: Parallel Algorithm and Application to the Argon Clusters
Yuri S. Nagornov, Ryosuke Akashi
Physica A: Statistical Mechanics and its Applications, 2019, 528, 121481.
DOI: 10.1016/j.physa.2019.121481
Learning Intermolecular Forces at Liquid-Vapor Interfaces
Samuel P. Niblett, Mirza Galib, David T. Limmer
2021.
Recursive Evaluation and Iterative Contraction of N-Body Equivariant Features
Jigyasa Nigam, Sergey Pozdnyakov, Michele Ceriotti
Journal of Chemical Physics, 2020, 153 (12), 121101.
DOI: 10.1063/5.0021116
Quantum-Accurate Magneto-Elastic Predictions with Classical Spin-Lattice Dynamics
Svetoslav Nikolov, Mitchell A. Wood, Attila Cangi, Jean-Bernard Maillet, Mihai-Cosmin Marinica, Aidan P. Thompson, Michael P. Desjarlais, Julien Tranchida
2021.
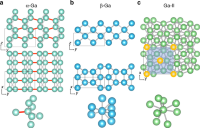
Ab Initio Phase Diagram and Nucleation of Gallium
Haiyang Niu, Luigi Bonati, Pablo M. Piaggi, Michele Parrinello
Nature Communications, 2020, 11 (1), 2654.
DOI: 10.1038/s41467-020-16372-9
The MLIP Package: Moment Tensor Potentials with MPI and Active Learning
Ivan S. Novikov, Konstantin Gubaev, Evgeny Podryabinkin, Alexander Shapeev
Machine Learning-Science and Technology, 2021, 2 (2), 025002.
DOI: 10.1088/2632-2153/abc9fe
Modeling H2O/Rutile-TiO2(110) Potential Energy Surfaces with Deep Networks
Stefan Oehmcke, Thomas Teusch, Thorben Petersen, Thorsten Kluener, Oliver Kramer
2020 International Joint Conference on Neural Networks (Ijcnn), 2020.
Catalytic Materials and Chemistry Development Using a Synergistic Combination of Machine Learning and Ab Initio Methods
Nilesh Varadan Orupattur, Samir H. Mushrif, Vinay Prasad
Computational Materials Science, 2020, 174, 109474.
DOI: 10.1016/j.commatsci.2019.109474
A Bin and Hash Method for Analyzing Reference Data and Descriptors in Machine Learning Potentials
Martin Leandro Paleico, Joerg Behler
Machine Learning-Science and Technology, 2021, 2 (3), 037001.
DOI: 10.1088/2632-2153/abe663
Machine Learning Assisted Free Energy Simulation of Solution–Phase and Enzyme Reactions
X Pan, R Van, E Epifanovsky, J Ho, J Huang, J Pu
2021.
A DFT Accurate Machine Learning Description of Molten ZnCl2 and Its Mixtures: 1. Potential Development and Properties Prediction of Molten ZnCl2
Gechuanqi Pan, Pin Chen, Hui Yan, Yutong Lu
Computational Materials Science, 2020, 185, 109955.
DOI: 10.1016/j.commatsci.2020.109955
A DFT Accurate Machine Learning Description of Molten ZnCl2 and Its Mixtures: 2. Potential Development and Properties Prediction of ZnCl2-NaCl-KCl Ternary Salt for CSP
Gechuanqi Pan, Jing Ding, Yunfei Du, Duu-Jong Lee, Yutong Lu
Computational Materials Science, 2021, 187, 110055.
DOI: 10.1016/j.commatsci.2020.110055
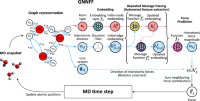
Accurate and Scalable Graph Neural Network Force Field and Molecular Dynamics with Direct Force Architecture
Cheol Woo Park, Mordechai Kornbluth, Jonathan Vandermause, Chris Wolverton, Boris Kozinsky, Jonathan P. Mailoa
Npj Computational Materials, 2021, 7 (1), 73.
DOI: 10.1038/s41524-021-00543-3
Accurate and Scalable Multi-Element Graph Neural Network Force Field and Molecular Dynamics with Direct Force Architecture
Cheol Woo Park, Mordechai Kornbluth, Jonathan Vandermause, Chris Wolverton, Jonathan P Mailoa
, 33.
A Fourier-Based Machine Learning Technique with Application in Engineering
Michael Peigney
International Journal for Numerical Methods in Engineering, 2021, 122 (3), 866–897.
DOI: 10.1002/nme.6565
Efficient Long-Range Convolutions for Point Clouds
Yifan Peng, Lin Lin, Lexing Ying, Leonardo Zepeda-Núñez
2020.
Simulations Meet Machine Learning in Structural Biology
Adrià Pérez, Gerard Martínez-Rosell, Gianni De Fabritiis
Current Opinion in Structural Biology, 2018, 49, 139–144.
DOI: 10/gdnsnp
Enhancing the Formation of Ionic Defects to Study the Ice Ih/XI Transition with Molecular Dynamics Simulations
Pablo M. Piaggi, Roberto Car
Molecular Physics, 2021.
DOI: 10.1080/00268976.2021.1916634
Phase Equilibrium of Water with Hexagonal and Cubic Ice Using the SCAN Functional
Pablo M. Piaggi, Athanassios Z. Panagiotopoulos, Pablo G. Debenedetti, Roberto Car
Journal of Chemical Theory and Computation, 2021, 17 (5), 3065–3077.
DOI: 10.1021/acs.jctc.1c00041
Machine Learning Force Fields: Recent Advances and Remaining Challenges
Igor Poltavsky, Alexandre Tkatchenko
Journal of Physical Chemistry Letters, 2021, 12 (28), 6551–6564.
DOI: 10.1021/acs.jpclett.1c01204
On Application of Deep Learning to Simplified Quantum-Classical Dynamics in Electronically Excited States
Evgeny Posenitskiy, Fernand Spiegelman, Didier Lemoine
Machine Learning-Science and Technology, 2021, 2 (3), 035039.
DOI: 10.1088/2632-2153/abfe3f
Atomistic Simulations of the Thermal Conductivity of Liquids
Marcello Puligheddu, Giulia Galli
Physical Review Materials, 2020, 4 (5), 053801.
DOI: 10.1103/PhysRevMaterials.4.053801
A Comprehensive Assessment of Empirical Potentials for Carbon Materials
Cheng Qian, Ben McLean, Daniel Hedman, Feng Ding
APL Materials, 2021, 9 (6).
DOI: 10.1063/5.0052870
OrbNet: Deep Learning for Quantum Chemistry Using Symmetry-Adapted Atomic-Orbital Features
Zhuoran Qiao, Matthew Welborn, Animashree Anandkumar, Frederick R. Manby, Thomas F. Miller
Journal of Chemical Physics, 2020, 153 (12), 124111.
DOI: 10.1063/5.0021955
Interaction Energy Prediction of Organic Molecules Using Deep Tensor Neural Network
Yuan Qi, Hong Ren, Hong Li, Ding-lin Zhang, Hong-qiang Cui, Jun-ben Weng, Guo-hui Li, Gui-yan Wang, Yan Li
Chinese Journal of Chemical Physics, 2021, 34 (1), 112–124.
DOI: 10.1063/1674-0068/cjcp2009163
Machine Learning of Atomic Forces from Quantum Mechanics: A Model Based on Pairwise Interatomic Forces
I Ramzan, L Kong, R A Bryce, N A Burton
, 39.
Unsupervised Learning of Atomic Environments from Simple Features
Wesley F. Reinhart
Computational Materials Science, 2021, 196, 110511.
DOI: 10.1016/j.commatsci.2021.110511
Halogen Bond Structure and Dynamics from Molecular Simulations
Richard C. Remsing, Michael L. Klein
Journal of Physical Chemistry B, 2019, 123 (29), 6266–6273.
DOI: 10.1021/acs.jpcb.9b04820
Machine Learning Kinetic Energy Functional for a One-Dimensional Periodic System
Hong-Bin Ren, Lei Wang, Xi Dai
Chinese Physics Letters, 2021, 38 (5), 050701.
DOI: 10.1088/0256-307X/38/5/050701
Spatial Density Neural Network Force Fields with First-Principles Level Accuracy and Application to Thermal Transport
Alejandro Rodriguez, Yinqiao Liu, Ming Hu
Physical Review B, 2020, 102 (3), 035203.
DOI: 10.1103/PhysRevB.102.035203
Biophysical Analysis of SARS-CoV-2 Transmission and Theranostic Development via N Protein Computational Characterization
Godfred O. Sabbih, Maame A. Korsah, Jaison Jeevanandam, Michael K. Danquah
Biotechnology Progress, 2021, 37 (2), e3096.
DOI: 10.1002/btpr.3096
Active Learning of Potential-Energy Surfaces of Weakly-Bound Complexes with Regression-Tree Ensembles
Yahya Saleh, Vishnu Sanjay, Armin Iske, Andrey Yachmenev, Jochen Küpper
2021.
Closing the Gap Between Modeling and Experiments in the Self-Assembly of Biomolecules at Interfaces and in Solution
Janani Sampath, Sarah Alamdari, Jim Pfaendtner
Chemistry of Materials, 2020, 32 (19), 8043–8059.
DOI: 10.1021/acs.chemmater.0c01891
Scalable Neural Networks for the Efficient Learning of Disordered Quantum Systems
N. Saraceni, S. Cantori, S. Pilati
Physical Review E, 2020, 102 (3).
DOI: 10.1103/physreve.102.033301
Molecular Force Fields with Gradient-Domain Machine Learning: Construction and Application to Dynamics of Small Molecules with Coupled Cluster Forces
Huziel E. Sauceda, Stefan Chmiela, Igor Poltavsky, Klaus-Robert Müller, Alexandre Tkatchenko
The Journal of Chemical Physics, 2019, 150 (11), 114102.
DOI: 10/ghqtd7
Kernel-Based Machine Learning for Efficient Simulations of Molecular Liquids
Christoph Scherer, Rene Scheid, Denis Andrienko, Tristan Bereau
Journal of Chemical Theory and Computation, 2020, 16 (5), 3194–3204.
DOI: 10.1021/acs.jctc.9b01256
From DFT to Machine Learning: Recent Approaches to Materials Science-a Review
Gabriel R. Schleder, Antonio C. M. Padilha, Carlos Mera Acosta, Marcio Costa, Adalberto Fazzio
Journal of Physics-Materials, 2019, 2 (3), 032001.
DOI: 10.1088/2515-7639/ab084b
Recent Advances and Applications of Machine Learning in Solid-State Materials Science
Jonathan Schmidt, Mario R. G. Marques, Silvana Botti, Miguel A. L. Marques
Npj Computational Materials, 2019, 5, 83.
DOI: 10.1038/s41524-019-0221-0
Committee Neural Network Potentials Control Generalization Errors and Enable Active Learning
Christoph Schran, Krystof Brezina, Ondrej Marsalek
Journal of Chemical Physics, 2020, 153 (10), 104105.
DOI: 10.1063/5.0016004
Transferability of Machine Learning Potentials: Protonated Water Neural Network Potential Applied to the Protonated Water Hexamer
Christoph Schran, Fabien Brieuc, Dominik Marx
Journal of Chemical Physics, 2021, 154 (5), 051101.
DOI: 10.1063/5.0035438
Schnet–a Deep Learning Architecture for Molecules and Materials
Kristof T. Schütt, Huziel E. Sauceda, P.-J. Kindermans, Alexandre Tkatchenko, K.-R. Müller
The Journal of Chemical Physics, 2018, 148 (24), 241722.
DOI: 10.1063/1.5019779
SchNetPack: A Deep Learning Toolbox For Atomistic Systems
K. T. Schütt, P. Kessel, M. Gastegger, K. Nicoli, A. Tkatchenko, K.-R. Müller
Journal of Chemical Theory and Computation, 2019, 15 (1), 448–455.
DOI: 10/gfrbqm
Differentiable Sampling of Molecular Geometries with Uncertainty-Based Adversarial Attacks
Daniel Schwalbe-Koda, Aik Rui Tan, Rafael Gómez-Bombarelli
2021.
Topology Automated Force-Field Interactions (TAFFI): A Framework for Developing Transferable Force-Fields
Bumjoon Seo, Zih-Yu Lin, Qiyuan Zhao, Michael A Webb, M Savoie
, 43.
Anharmonic Raman Spectra Simulation of Crystals from Deep Neural Networks
Honghui Shang, Haidi Wang
Aip Advances, 2021, 11 (3), 035105.
DOI: 10.1063/5.0040190
Modelling Bulk Electrolytes and Electrolyte Interfaces with Atomistic Machine Learning
Yunqi Shao, Lisanne Knijff, Florian M. Dietrich, Kersti Hermansson, Chao Zhang
Batteries \& Supercaps, 2021, 4 (4), 585–595.
DOI: 10.1002/batt.202000262
PiNN: A Python Library for Building Atomic Neural Networks of Molecules and Materials
Yunqi Shao, Matti Hellstrom, Pavlin D. Mitev, Lisanne Knijff, Chao Zhang
Journal of Chemical Information and Modeling, 2020, 60 (3), 1184–1193.
DOI: 10.1021/acs.jcim.9b00994
Elinvar Effect in Beta-Ti Simulated by on-the-Fly Trained Moment Tensor Potential
Alexander Shapeev, Evgeny Podryabinkin, Konstantin Gubaev, Ferenc Tasnadi, Igor A. Abrikosov
New Journal of Physics, 2020, 22 (11), 113005.
DOI: 10.1088/1367-2630/abc392
PFNN: A Penalty-Free Neural Network Method for Solving a Class of Second-Order Boundary-Value Problems on Complex Geometries
Hailong Sheng, Chao Yang
Journal of Computational Physics, 2021, 428, 110085.
DOI: 10.1016/j.jcp.2020.110085
Quantum Trajectory Mean-Field Method for Nonadiabatic Dynamics in Photochemistry
Lin Shen, Diandong Tang, Binbin Xie, Wei-Hai Fang
Journal of Physical Chemistry A, 2019, 123 (34), 7337–7350.
DOI: 10.1021/acs.jpca.9b03480
Application of Genetic Algorithm in the Global Structure Optimization of Catalytic System
Xiangcheng Shi, Zhijian Zhao, Jinlong Gong
Huagong Xuebao/CIESC Journal, 2021, 72 (1), 27–41.
DOI: 10.11949/0438-1157.20201037
Learning Gradient Fields for Molecular Conformation Generation
Chence Shi, Shitong Luo, Minkai Xu, Jian Tang
2021.
Computational and Training Requirements for Interatomic Potential Based on Artificial Neural Network for Estimating Low Thermal Conductivity of Silver Chalcogenides
Kohei Shimamura, Yusuke Takeshita, Shogo Fukushima, Akihide Koura, Fuyuki Shimojo
Journal of Chemical Physics, 2020, 153 (23), 234301.
DOI: 10.1063/5.0027058
Estimating Thermal Conductivity of α-Ag2Se Using ANN Potential with Chebyshev Descriptor
Kohei Shimamura, Yusuke Takeshita, Shogo Fukushima, Akihide Koura, Fuyuki Shimojo
Chemical Physics Letters, 2021, 778, 138748.
DOI: 10/gj42cx
Guidelines for Creating Artificial Neural Network Empirical Interatomic Potential from First-Principles Molecular Dynamics Data under Specific Conditions and Its Application to Alpha-Ag2Se
Kohei Shimamura, Shogo Fukushima, Akihide Koura, Fuyuki Shimojo, Masaaki Misawa, Rajiv K. Kalia, Aiichiro Nakano, Priya Vashishta, Takashi Matsubara, Shigenori Tanaka
Journal of Chemical Physics, 2019, 151 (12), 124303.
DOI: 10.1063/1.5116420
Water Dipole and Quadrupole Moment Contributions to the Ion Hydration Free Energy by the Deep Neural Network Trained with Ab Initio Molecular Dynamics Data
Yu Shi, Carrie C Doyle, Thomas L Beck
, 20.
Wavelet Scattering Networks for Atomistic Systems with Extrapolation of Material Properties
Paul Sinz, Michael W. Swift, Xavier Brumwell, Jialin Liu, Kwang Jin Kim, Yue Qi, Matthew Hirn
Journal of Chemical Physics, 2020, 153 (8), 084109.
DOI: 10.1063/5.0016020
Experimentally Driven Automated Machine-Learned Interatomic Potential for a Refractory Oxide
Ganesh Sivaraman, Leighanne Gallington, Anand Narayanan Krishnamoorthy, Marius Stan, Gábor Csányi, Álvaro Vázquez-Mayagoitia, Chris J. Benmore
Physical Review Letters, 2021, 126 (15), 156002.
DOI: 10/gkx66f
The ANI-1ccx and ANI-1x Data Sets, Coupled-Cluster and Density Functional Theory Properties for Molecules
Justin S. Smith, Roman Zubatyuk, Benjamin Nebgen, Nicholas Lubbers, Kipton Barros, Adrian E. Roitberg, Olexandr Isayev, Sergei Tretiak
Scientific Data, 2020, 7 (1), 134.
DOI: 10/gh48xw
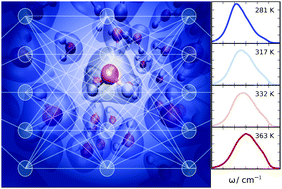
Raman Spectrum and Polarizability of Liquid Water from Deep Neural Networks
Grace M. Sommers, Marcos F. Calegari Andrade, Linfeng Zhang, Han Wang, Roberto Car
Physical Chemistry Chemical Physics, 2020, 22 (19), 10592–10602.
DOI: 10.1039/d0cp01893g
Machine Learning for Metallurgy II. A Neural-Network Potential for Magnesium
Markus Stricker, Binglun Yin, Eleanor Mak, W. A. Curtin
Physical Review Materials, 2020, 4 (10).
DOI: 10.1103/physrevmaterials.4.103602
Toward Exascale Design of Soft Mesoscale Materials
S Succi, G Amati, F Bonaccorso, M Lauricella - Journal of …, undefined 2020
Elsevier, 2020.
Gaussian Process Model of 51-Dimensional Potential Energy Surface for Protonated Imidazole Dimer
Hiroki Sugisawa, Tomonori Ida, R. Krems
Journal of Chemical Physics, 2020, 153 (11), 114101.
DOI: 10.1063/5.0023492
TeaNet: Universal Neural Network Interatomic Potential Inspired by Iterative Electronic Relaxations
So Takamoto, Satoshi Izumi, Ju Li
2019.
Interatomic Potential in a Simple Dense Neural Network Representation
Ka-Ming Tam, Nicholas Walker, Samuel Kellar, Mark Jarrell
2019.
Prediction of Formation Energies of Large-Scale Disordered Systems via Active-Learning-Based Executions of Ab Initio Local-Energy Calculations: A Case Study on a Fe Random Grain Boundary Model with Millions of Atoms
Tomoyuki Tamura, Masayuki Karasuyama
Physical Review Materials, 2020, 4 (11).
DOI: 10.1103/physrevmaterials.4.113602
ChebNet: Efficient and Stable Constructions of Deep Neural Networks with Rectified Power Units Using Chebyshev Approximations
Shanshan Tang, Bo Li, Haijun Yu
2019.
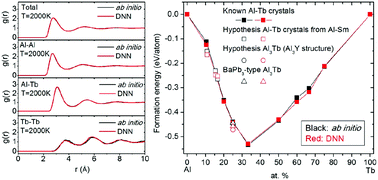
Development of Interatomic Potential for Al-Tb Alloys Using a Deep Neural Network Learning Method
L. Tang, Z. J. Yang, T. Q. Wen, K. M. Ho, M. J. Kramer, C. Z. Wang
Physical Chemistry Chemical Physics, 2020, 22 (33), 18467–18479.
DOI: 10.1039/d0cp01689f
Short- and Medium-Range Orders in Al90Tb10 Glass and Their Relation to the Structures of Competing Crystalline Phases
L. Tang, Z. J. Yang, T. Q. Wen, K. M. Ho, M. J. Kramer, C. Z. Wang
Acta Materialia, 2021, 204, 116513.
DOI: 10.1016/j.actamat.2020.116513
Machine Learning and Molecular Design of Self-Assembling Pi-Conjugated Oligopeptides
Bryce A. Thurston, Andrew L. Ferguson
Molecular Simulation, 2018, 44 (11), 930–945.
DOI: 10.1080/08927022.2018.1469754
The Repetitive Local Sampling and the Local Distribution Theory
Pu Tian
, 32.
Combining Machine Learning Potential and Structure Prediction for Accelerated Materials Design and Discovery
Qunchao Tong, Pengyue Gao, Hanyu Liu, Yu Xie, Jian Lv, Yanchao Wang, Jijun Zhao
Journal of Physical Chemistry Letters, 2020, 11 (20), 8710–8720.
DOI: 10.1021/acs.jpclett.0c02357
Machine Learning Metadynamics Simulation of Reconstructive Phase Transition
Qunchao Tong, Xiaoshan Luo, Adebayo A. Adeleke, Pengyue Gao, Yu Xie, Hanyu Liu, Quan Li, Yanchao Wang, Jian Lv, Yansun Yao, Yanming Ma
Physical Review B, 2021, 103 (5), 054107.
DOI: 10/gmf5zv
Geometric Prediction: Moving Beyond Scalars
Raphael J. L. Townshend, Brent Townshend, Stephan Eismann, Ron O. Dror
2020.
Transferrable End-to-End Learning for Protein Interface Prediction
Raphael JL Townshend, Rishi Bedi, Ron O. Dror
2018.
A Machine Learning Based Deep Potential for Seeking the Low-Lying Candidates of Al Clusters
P. Tuo, X. B. Ye, B. C. Pan
Journal of Chemical Physics, 2020, 152 (11), 114105.
DOI: 10.1063/5.0001491
PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges
Oliver T. Unke, Markus Meuwly
Journal of Chemical Theory and Computation, 2019, 15 (6), 3678–3693.
DOI: 10.1021/acs.jctc.9b00181
SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects
Oliver T. Unke, Stefan Chmiela, Michael Gastegger, Kristof T. Schütt, Huziel E. Sauceda, Klaus-Robert Müller
2021.
Active Learning of Reactive Bayesian Force Fields: Application to Heterogeneous Hydrogen-Platinum Catalysis Dynamics
J Vandermause, Y Xie, JS Lim, CJ Owen - arXiv preprint arXiv …, undefined 2021
arxiv.org, 2021.
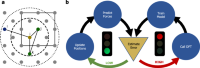
On-the-Fly Active Learning of Interpretable Bayesian Force Fields for Atomistic Rare Events
Jonathan Vandermause, Steven B. Torrisi, Simon Batzner, Yu Xie, Lixin Sun, Alexie M. Kolpak, Boris Kozinsky
Npj Computational Materials, 2020, 6 (1), 20.
DOI: 10.1038/s41524-020-0283-z
On-the-Fly Bayesian Active Learning of Interpretable Force-Fields for Atomistic Rare Events
J Vandermause, SB Torrisi, S Batzner
projects.iq.harvard.edu, 2019.
Challenges for Machine Learning Force Fields in Reproducing Potential Energy Surfaces of Flexible Molecules
Valentin Vassilev-Galindo, Gregory Fonseca, Igor Poltavsky, Alexandre Tkatchenko
Journal of Chemical Physics, 2021, 154 (9), 094119.
DOI: 10.1063/5.0038516
Bayesian Machine Learning Approach to the Quantification of Uncertainties on Ab Initio Potential Energy Surfaces
S. Venturi, R. L. Jaffe, M. Panesi
Journal of Physical Chemistry A, 2020, 124 (25), 5129–5146.
DOI: 10.1021/acs.jpca.0c02395
Molecular Modeling Investigations of Sorption and Diffusion of Small Molecules in Glassy Polymers
Niki Vergadou, Doros N. Theodorou
Membranes, 2019, 9 (8), 98.
DOI: 10.3390/membranes9080098
Faster Exact Exchange in Periodic Systems Using Single-Precision Arithmetic
John Vinson
Journal of Chemical Physics, 2020, 153 (20), 204106.
DOI: 10.1063/5.0030493
Combining the Fragmentation Approach and Neural Network Potential Energy Surfaces of Fragments for Accurate Calculation of Protein Energy
Zhilong Wang, Yanqiang Han, Jinjin Li, Xiao He
Journal of Physical Chemistry B, 2020, 124 (15), 3027–3035.
DOI: 10.1021/acs.jpcb.0c01370
Complex Reaction Network Thermodynamic and Kinetic Autoconstruction Based on \emphAb Initio Statistical Mechanics: A Case Study of O \textsubscript2 Activation on Ag \textsubscript4 Clusters
Weiqi Wang, Xiangyue Liu, Jesús Pérez-Ríos
The Journal of Physical Chemistry A, 2021, 125 (25), 5670–5680.
DOI: 10/gmfw5m
Crystal Structure Prediction of Binary Alloys via Deep Potential
Haidi Wang, Yuzhi Zhang, Linfeng Zhang, Han Wang
Frontiers in Chemistry, 2020, 8, 589795.
DOI: 10.3389/fchem.2020.589795
Deep Learning Inter-Atomic Potential Model for Accurate Irradiation Damage Simulations
Hao Wang, Xun Guo, Linfeng Zhang, Han Wang, Jianming Xue
Applied Physics Letters, 2019, 114 (24), 244101.
DOI: 10.1063/1.5098061
Deep-Learning Interatomic Potential for Irradiation Damage Simulations in MoS2 with Ab Initial Accuracy
Hao Wang, Xun Guo, Jianming Xue
2020.
DeePMD-Kit: A Deep Learning Package for Many-Body Potential Energy Representation and Molecular Dynamics
Han Wang, Linfeng Zhang, Jiequn Han, Weinan E
Computer Physics Communications, 2018, 228, 178–184.
DOI: 10.1016/j.cpc.2018.03.016
Differentiable Molecular Simulations for Control and Learning
Wujie Wang, Simon Axelrod, Rafael Gómez-Bombarelli
2020.
Electronically Driven 1D Cooperative Diffusion in a Simple Cubic Crystal
Yong Wang, Junjie Wang, Andreas Hermann, Cong Liu, Hao Gao, Erio Tosatti, Hui-Tian Wang, Dingyu Xing, Jian Sun
Physical Review X, 2021, 11 (1), 011006.
DOI: 10.1103/PhysRevX.11.011006
Ensemble Learning of Coarse-Grained Molecular Dynamics Force Fields with a Kernel Approach
Jiang Wang, Stefan Chmiela, Klaus-Robert Mueller, Frank Noe, Cecilia Clementi
Journal of Chemical Physics, 2020, 152 (19).
DOI: 10.1063/5.0007276
An Extendible, Graph-Neural-Network-Based Approach for Accurate Force Field Development of Large Flexible Organic Molecules
Xufei Wang, Yuanda Xu, Han Zheng, Kuang Yu
arxiv.org, 2021.
Machine Learning of Coarse-Grained Molecular Dynamics Force Fields
Jiang Wang, Simon Olsson, Christoph Wehmeyer, Adria Perez, Nicholas E. Charron, Gianni de Fabritiis, Frank Noe, Cecilia Clementi
Acs Central Science, 2019, 5 (5), 755–767.
DOI: 10.1021/acscentsci.8b00913
Multi-Body Effects in a Coarse-Grained Protein Force Field
Jiang Wang, Nicholas Charron, Brooke Husic, Simon Olsson, Frank Noé, Cecilia Clementi
Journal of Chemical Physics, 2021, 154 (16).
DOI: 10.1063/5.0041022
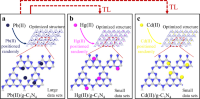
Predicting Adsorption Ability of Adsorbents at Arbitrary Sites for Pollutants Using Deep Transfer Learning
Zhilong Wang, Haikuo Zhang, Jiahao Ren, Xirong Lin, Tianli Han, Jinyun Liu, Jinjin Li
Npj Computational Materials, 2021, 7 (1), 19.
DOI: 10.1038/s41524-021-00494-9
Symmetry-Adapted Graph Neural Networks for Constructing Molecular Dynamics Force Fields
Zun Wang, Chong Wang, Sibo Zhao, Shiqiao Du, Yong Xu, Bing-Lin Gu, Wenhui Duan
2021.
Integrating Machine Learning with Physics-Based Modeling
E Weinan, Jiequn Han, Zhang Linfeng
2020.
Properties of Alpha-Brass Nanoparticles. 1. Neural Network Potential Energy Surface
Jan Weinreich, Anton Roemer, Martin Leandro Paleico, Joerg Behler
Journal of Physical Chemistry C, 2020, 124 (23), 12682–12695.
DOI: 10.1021/acs.jpcc.0c00559
Development of a Deep Machine Learning Interatomic Potential for Metalloid-Containing Pd-Si Compounds
Tongqi Wen, Cai-Zhuang Wang, M. J. Kramer, Yang Sun, Beilin Ye, Haidi Wang, Xueyuan Liu, Chao Zhang, Feng Zhang, Kai-Ming Ho, Nan Wang
Physical Review B, 2019, 100 (17), 174101.
DOI: 10.1103/PhysRevB.100.174101
Combining SchNet and SHARC: The SchNarc Machine Learning Approach for Excited-State Dynamics
Julia Westermayr, Michael Gastegger, Philipp Marquetand
Journal of Physical Chemistry Letters, 2020, 11 (10), 3828–3834.
DOI: 10.1021/acs.jpclett.0c00527
Machine Learning and Excited-State Molecular Dynamics
Julia Westermayr, Philipp Marquetand
Machine Learning: Science and Technology, 2020, 1 (4), 043001.
DOI: 10/gksxpp
Atom-Density Representations for Machine Learning
Michael J. Willatt, Flix Musil, Michele Ceriotti
Journal of Chemical Physics, 2019, 150 (15), 154110.
DOI: 10.1063/1.5090481
Feature Optimization for Atomistic Machine Learning Yields A Data-Driven Construction of the Periodic Table of the Elements
Michael J. Willatt, Félix Musil, Michele Ceriotti
Physical Chemistry Chemical Physics, 2018, 20 (47), 29661–29668.
DOI: 10/gfz26d
Targeted Free Energy Estimation via Learned Mappings
Peter Wirnsberger, Andrew J. Ballard, George Papamakarios, Stuart Abercrombie, Sebastien Racaniere, Alexander Pritzel, Danilo Jimenez Rezende, Charles Blundell
Journal of Chemical Physics, 2020, 153 (14), 144112.
DOI: 10.1063/5.0018903
Active Learning Approach to Optimization of Experimental Control
Y Wu, Z Meng, K Wen, C Mi, Zhang J
iopscience.iop.org.
Deep Learning of Accurate Force Field of Ferroelectric HfO2
Jing Wu, Yuzhi Zhang, Linfeng Zhang, Shi Liu
Physical Review B, 2021, 103 (2), 024108.
DOI: 10.1103/PhysRevB.103.024108
Deep Learning of Accurate Force Field of Ferroelectric HfO2
Jing Wu, Yuzhi Zhang, Linfeng Zhang, Shi Liu
Physical Review B, 2021, 103 (2), 024108.
DOI: 10.1103/PhysRevB.103.024108
Modeling of Metal Nanoparticles: Development of Neural-Network Interatomic Potential Inspired by Features of the Modified Embedded-Atom Method
Feifeng Wu, Hang Min, Yanwei Wen, Rong Chen, Yunkun Zhao, Mike Ford, Bin Shan
Physical Review B, 2020, 102 (14), 144107.
DOI: 10.1103/PhysRevB.102.144107
High-Throughput Study of Lattice Thermal Conductivity in Binary Rocksalt and Zinc Blende Compounds Including Higher-Order Anharmonicity
Yi Xia, Vinay Hegde, Koushik Pal, Xia Hua, Dale Gaines, Shane Patel, Jiangang He, Muratahan Aykol, Chris Wolverton
Physical Review X, 2020, 10 (4), 041029.
DOI: 10.1103/PhysRevX.10.041029
Ab-Initio Study of Interacting Fermions at Finite Temperature with Neural Canonical Transformation
Hao Xie, Linfeng Zhang, Lei Wang
arxiv.org, 2021.
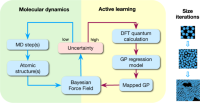
Bayesian Force Fields from Active Learning for Simulation of Inter-Dimensional Transformation of Stanene
Yu Xie, Jonathan Vandermause, Lixin Sun, Andrea Cepellotti, Boris Kozinsky
Npj Computational Materials, 2021, 7 (1), 40.
DOI: 10.1038/s41524-021-00510-y
Graph Dynamical Networks for Unsupervised Learning of Atomic Scale Dynamics in Materials
Tian Xie, Arthur France-Lanord, Yanming Wang, Yang Shao-Horn, Jeffrey C. Grossman
Nature Communications, 2019, 10, 2667.
DOI: 10.1038/s41467-019-10663-6
Automated Construction of Neural Network Potential Energy Surface: The Enhanced Self-Organizing Incremental Neural Network Deep Potential Method
Mingyuan Xu, Tong Zhu, John Z H Zhang
, 18.
Automatically Constructed Neural Network Potentials for Molecular Dynamics Simulation of Zinc Proteins
Mingyuan Xu, Tong Zhu, John Z. H. Zhang
Frontiers in Chemistry, 2021, 9, 692200.
DOI: 10.3389/fchem.2021.692200
A Deep-Learning Potential for Crystalline and Amorphous Li-Si Alloys
Nan Xu, Yao Shi, Yi He, Qing Shao
Journal of Physical Chemistry C, 2020, 124 (30), 16278–16288.
DOI: 10.1021/acs.jpcc.0c03333
Ab Initio Molecular Dynamics Simulation of Zinc Metalloproteins with Enhanced Self-Organizing Incremental High Dimensional Neural Network
Mingyuan Xu, Tong Zhu, John Z H Zhang
, 27.
Isotope Effects in Molecular Structures and Electronic Properties of Liquid Water via Deep Potential Molecular Dynamics Based on the SCAN Functional
Jianhang Xu, Chunyi Zhang, Linfeng Zhang, Mohan Chen, Biswajit Santra, Xifan Wu
Physical Review B, 2020, 102 (21), 214113.
DOI: 10.1103/PhysRevB.102.214113
Isotope Effects in Molecular Structures and Electronic Properties of Liquid Water via Deep Potential Molecular Dynamics Based on the SCAN Functional
Jianhang Xu, Chunyi Zhang, Linfeng Zhang, Mohan Chen, Biswajit Santra, Xifan Wu
Physical Review B, 2020, 102 (21), 214113.
DOI: 10.1103/PhysRevB.102.214113
Molecular Dynamics Simulation of Zinc Ion in Water with an Ab Initio Based Neural Network Potential
Mingyuan Xu, Tong Zhu, John Z. H. Zhang
Journal of Physical Chemistry A, 2019, 123 (30), 6587–6595.
DOI: 10.1021/acs.jpca.9b04087
De Novo Molecule Design through Molecular Generative Model Conditioned by 3D Information of Protein Binding Sites
Mingyuan Xu, Ting Ran, Hongming Chen
, 25.
Optimizing Training Data Set for the Machine Learning Potential of Li-Si Alloys via Structural Similarity-Based Screening
Nan Xu, Chen Li, Yao Shi, Qing Shao, Yi He
arxiv.org, 2021.
Perspective on Computational Reaction Prediction Using Machine Learning Methods in Heterogeneous Catalysis
Jiayan Xu, Xiao-Ming Cao, P. Hu
Physical Chemistry Chemical Physics, 2021, 23 (19), 11155–11179.
DOI: 10.1039/d1cp01349a
Using Metadynamics to Build Neural Network Potentials for Reactive Events: The Case of Urea Decomposition in Water
M Yang, L Bonati, D Polino, Parrinello M
Elsevier, 2021.
Construction of a Neural Network Energy Function for Protein Physics
Huan Yang, Zhaoping Xiong, Francesco Zonta
2021.
DOI: 10.1101/2021.04.26.441401
Role of Water in the Reaction Mechanism and Endo/Exo Selectivity of 1,3-Dipolar Cycloadditions Elucidated by Quantum Chemistry and Machine Learning
Xin Yang, Jun Zou, Yifei Wang, Ying Xue, Shengyong Yang
Chemistry-a European Journal, 2019, 25 (35), 8289–8303.
DOI: 10.1002/chem.201900617
Active Learning Algorithm for Computational Physics
J Yao, Y Wu, J Koo, B Yan, Zhai H
APS, 2020, 2 (1), 13287.
DOI: 10.1103/physrevresearch.2.013287
Nuclear Quantum Effect and Its Temperature Dependence in Liquid Water from Random Phase Approximation via Artificial Neural Network
Yi Yao, Yosuke Kanai
The Journal of Physical Chemistry Letters, 2021, 12 (27), 6354–6362.
DOI: 10/gk5v27
Atomic Energy Mapping of Neural Network Potential
Dongsun Yoo, Kyuhyun Lee, Wonseok Jeong, Dongheon Lee, Satoshi Watanabe, Seungwu Han
Physical Review Materials, 2019, 3 (9), 093802.
DOI: 10.1103/PhysRevMaterials.3.093802
A Transferable Active-Learning Strategy for Reactive Molecular Force Fields
Tom A. Young, Tristan Johnston-Wood, Volker L. Deringer, Fernanda Duarte
Chemical Science, 2021.
DOI: 10.1039/d1sc01825f
When Do Short-Range Atomistic Machine-Learning Models Fall Short?
Shuwen Yue, Maria Carolina Muniz, Marcos F. Calegari Andrade, Linfeng Zhang, Roberto Car, Athanassios Z. Panagiotopoulos
The Journal of Chemical Physics, 2021, 154 (3), 034111.
DOI: 10/gkcq6f
Explore the Chemical Space of Linear Alkanes Pyrolysis via Deep Potential Generator
J Zeng, L Zhang, H Wang, T Zhu
2020.
Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/Molecular Mechanical Simulations of Chemical Reactions in Solution
J Zeng, TJ Giese, Ş Ekesan, DM York
2021.
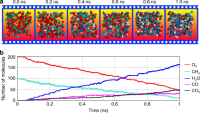
Complex Reaction Processes in Combustion Unraveled by Neural Network-Based Molecular Dynamics Simulation
Jinzhe Zeng, Liqun Cao, Mingyuan Xu, Tong Zhu, John Z. H. Zhang
Nature Communications, 2020, 11 (1), 5713.
DOI: 10.1038/s41467-020-19497-z
Complex Reaction Processes in Combustion Unraveled by Neural Network-Based Molecular Dynamics Simulation
Jinzhe Zeng, Liqun Cao, Mingyuan Xu, Tong Zhu, John Z. H. Zhang
Nature Communications, 2020, 11 (1), 5713.
DOI: 10.1038/s41467-020-19497-z
Exploring the Chemical Space of Linear Alkane Pyrolysis via Deep Potential GENerator
Jinzhe Zeng, Linfeng Zhang, Han Wang, Tong Zhu
Energy \& Fuels, 2021, 35 (1), 762–769.
DOI: 10.1021/acs.energyfuels.0c03211
Neural Network Based in Silico Simulation of Combustion Reactions
Jinzhe Zeng, Liqun Cao, Mingyuan Xu, Tong Zhu, John ZH Zhang
arxiv.org, 2019.
Deep Density: Circumventing the Kohn-Sham Equations via Symmetry Preserving Neural Networks
Leonardo Zepeda-Núñez, Yixiao Chen, Jiefu Zhang, Weile Jia, Linfeng Zhang, Lin Lin
Elsevier, 2019.
Active Learning of Many-Body Configuration Space: Application to the Cs+-Water MB-Nrg Potential Energy Function as a Case Study
Yaoguang Zhai, Alessandro Caruso, Sicun Gao, Francesco Paesani
Journal of Chemical Physics, 2020, 152 (14), 144103.
DOI: 10.1063/5.0002162
BubbleNet: Inferring Micro-Bubble Dynamics with Semi-Physics-Informed Deep Learning
Hanfeng Zhai, Guohui Hu
2021.
Machine Learning for Multi-Scale Molecular Modeling: Theories, Algorithms, and Applications
L Zhang
2020.
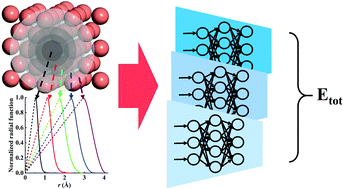
Accelerating Atomistic Simulations with Piecewise Machine-Learned Ab Initio Potentials at a Classical Force Field-like Cost
Yaolong Zhang, Ce Hu, Bin Jiang
Physical Chemistry Chemical Physics, 2021, 23 (3), 1815–1821.
DOI: 10.1039/d0cp05089j
Active Learning of Uniformly Accurate Interatomic Potentials for Materials Simulation
Linfeng Zhang, De-Ye Lin, Han Wang, Roberto Car, Weinan E
Physical Review Materials, 2019, 3 (2), 023804.
DOI: 10.1103/PhysRevMaterials.3.023804
Adaptive Coupling of a Deep Neural Network Potential to a Classical Force Field
Linfeng Zhang, Han Wang, Weinan E
The Journal of chemical physics, 2018, 149 (15), 154107.
DOI: 10.1063/1.5042714
Anomalous Phase Separation and Hidden Coarsening of Super-Clusters in the Falicov-Kimball Model
Sheng Zhang, Puhan Zhang, Gia-Wei Chern
2021.
Arrested Phase Separation in Double-Exchange Models: Machine-Learning Enabled Large-Scale Simulation
Puhan Zhang, Gia-Wei Chern
2021.
Bridging the Gap between Direct Dynamics and Globally Accurate Reactive Potential Energy Surfaces Using Neural Networks
Yaolong Zhang, Xueyao Zhou, Bin Jiang
Journal of Physical Chemistry Letters, 2019, 10 (6), 1185–1191.
DOI: 10.1021/acs.jpclett.9b00085
Crystallization of the P3Sn4 Phase upon Cooling P2Sn5 Liquid by Molecular Dynamics Simulation Using a Machine Learning Interatomic Potential
Chao Zhang, Yang Sun, Hai-Di Wang, Feng Zhang, Tong-Qi Wen, Kai-Ming Ho, Cai-Zhuang Wang
Journal of Physical Chemistry C, 2021, 125 (5), 3127–3133.
DOI: 10.1021/acs.jpcc.0c08873
DeePCG: Constructing Coarse-Grained Models via Deep Neural Networks
Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, Weinan E. Weinan
2018, 149 (3).
DOI: 10.1063/1.5027645
Deep Neural Network for the Dielectric Response of Insulators
Linfeng Zhang, Mohan Chen, Xifan Wu, Han Wang, E. Weinan, Roberto Car
Physical Review B, 2020, 102 (4), 041121.
DOI: 10.1103/PhysRevB.102.041121
Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics
Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, E. Weinan
Physical Review Letters, 2018, 120 (14), 143001.
DOI: 10.1103/PhysRevLett.120.143001
DP-GEN: A Concurrent Learning Platform for the Generation of Reliable Deep Learning Based Potential Energy Models
Yuzhi Zhang, Haidi Wang, Weijie Chen, Jinzhe Zeng, Linfeng Zhang, Han Wang, E. Weinan
Computer Physics Communications, 2020, 253, 107206.
DOI: 10.1016/j.cpc.2020.107206
Efficient and Accurate Simulations of Vibrational and Electronic Spectra with Symmetry-Preserving Neural Network Models for Tensorial Properties
Yaolong Zhang, Sheng Ye, Jinxiao Zhang, Ce Hu, Jun Jiang, Bin Jiang
The Journal of Physical Chemistry B, 2020, 124 (33), 7284–7290.
DOI: 10.1021/acs.jpcb.0c06926
Embedded Atom Neural Network Potentials: Efficient and Accurate Machine Learning with a Physically Inspired Representation
Yaolong Zhang, Ce Hu, Bin Jiang
Journal of Physical Chemistry Letters, 2019, 10 (17), 4962–4967.
DOI: 10.1021/acs.jpclett.9b02037
Embedded Atom Neural Network Potentials: Efficient and Accurate Machine Learning with a Physically Inspired Representation
Yaolong Zhang, Ce Hu, Bin Jiang
Journal of Physical Chemistry Letters, 2019, 10 (17), 4962–4967.
DOI: 10.1021/acs.jpclett.9b02037
End-to-End Symmetry Preserving Inter-Atomic Potential Energy Model for Finite and Extended Systems
Linfeng Zhang, Jiequn Han, Han Wang, Wissam A. Saidi, Roberto Car, Weinan E
2018.
DOI: arXiv:1805.09003
Global Optimization of Chemical Cluster Structures: Methods, Applications, and Challenges
Jun Zhang, Vassiliki-Alexandra Glezakou
International Journal of Quantum Chemistry, 2021, 121 (7), e26553.
DOI: 10.1002/qua.26553
Isotope Effects in X-Ray Absorption Spectra of Liquid Water
Chunyi Zhang, Linfeng Zhang, Jianhang Xu, Fujie Tang, Biswajit Santra, Xifan Wu
Physical Review B, 2020, 102 (11), 115155.
DOI: 10.1103/PhysRevB.102.115155
A Linear Frequency Principle Model to Understand the Absence of Overfitting in Neural Networks
Yaoyu Zhang, Tao Luo, Zheng Ma, Zhi-Qin John Xu
Chinese Physics Letters, 2021, 38 (3), 038701.
DOI: 10.1088/0256-307X/38/3/038701
Machine Learning Dynamics of Phase Separation in Correlated Electron Magnets
Puhan Zhang, Preetha Saha, Gia-Wei Chern
2020.
Molecular CT: Unifying Geometry and Representation Learning for Molecules at Different Scales
Jun Zhang, Yaqiang Zhou, Yao-Kun Lei, Yi Isaac Yang, Yi Qin Gao
, 14.
Monge-Amp\$\textbackslash backslash\$ere Flow for Generative Modeling
Linfeng Zhang, Lei Wang
2018.
A Perspective on Deep Learning for Molecular Modeling and Simulations
Jun Zhang, Yao-Kun Lei, Zhen hZang, Junhan Chang, Maodong Li, Xu Han, Lijiang Yang, Yi Isaac Yang, Yi Qin Gao
Journal of Physical Chemistry A, 2020, 124 (34), 6745–6763.
DOI: 10.1021/acs.jpca.0c04473
Phase Diagram of a Deep Potential Water Model
Linfeng Zhang, Han Wang, Roberto Car, E. Weinan
Physical Review Letters, 2021, 126 (23), 236001.
DOI: 10.1103/PhysRevLett.126.236001
Reinforced Dynamics for Enhanced Sampling in Large Atomic and Molecular Systems
Linfeng Zhang, Han Wang, Weinan E
The Journal of chemical physics, 2018, 148 (12), 124113.
DOI: 10.1063/1.5019675
Reinforcement Learning for Multi-Scale Molecular Modeling
Jun Zhang, Yao-Kun Lei, Yi Isaac Yang, Yi Qin Gao
, 26.
A Type of Generalization Error Induced by Initialization in Deep Neural Networks
Yaoyu Zhang, Zhi-Qin John Xu, Tao Luo, Zheng Ma
, 21.
Warm Dense Matter Simulation via Electron Temperature Dependent Deep Potential Molecular Dynamics
Yuzhi Zhang, Chang Gao, Qianrui Liu, Linfeng Zhang, Han Wang, Mohan Chen
Physics of Plasmas, 2020, 27 (12), 122704.
DOI: 10.1063/5.0023265
Learning the Physics of Pattern Formation from Images
Hongbo Zhao, Brian D. Storey, Richard D. Braatz, Martin Z. Bazant
Physical Review Letters, 2020, 124 (6), 060201.
DOI: 10.1103/PhysRevLett.124.060201
Theoretical Prediction on the Redox Potentials of Rare-Earth Ions by Deep Potentials
Jia Zhao, Wenshuo Liang, Guimin Lu
Ionics, 2021, 27 (5), 2079–2088.
DOI: 10/gmfwvw
Retention and Recycling of Deuterium in Liquid Lithium-Tin Slab Studied by First-Principles Molecular Dynamics
Daye Zheng, Zhen-Xiong Shen, Mohan Chen, Xinguo Ren, Lixin He
Journal of Nuclear Materials, 2021, 543, 152542.
DOI: 10.1016/j.jnucmat.2020.152542

Atomic-State-Dependent Screening Model for Hot and Warm Dense Plasmas
Fuyang Zhou, Yizhi Qu, Junwen Gao, Yulong Ma, Yong Wu, Jianguo Wang
Communications Physics, 2021, 4 (1), 148.
DOI: 10.1038/s42005-021-00652-x
Frame-Independent Vector-Cloud Neural Network for Nonlocal Constitutive Modelling on Arbitrary Grids
Xu-Hui Zhou, Jiequn Han, Heng Xiao
2021.
Structure and Dynamics of Supercooled Liquid Ge \textsubscript2 Sb \textsubscript2 Te \textsubscript5 from Machine‐Learning‐Driven Simulations
Yu-Xing Zhou, Han-Yi Zhang, Volker L. Deringer, Wei Zhang
physica status solidi (RRL) – Rapid Research Letters, 2021, 15 (3), 2000403.
DOI: 10/gmf6g6
Discriminating High-Pressure Water Phases Using Rare-Event Determined Ionic Dynamical Properties*
Lin Zhuan, Qijun Ye, Ding Pan, Xin-Zheng Li
Chinese Physics Letters, 2020, 37 (4), 043101.
DOI: 10.1088/0256-307X/37/4/043101
Development of Multimodal Machine Learning Potentials: Toward a Physics-Aware Artificial Intelligence
Tetiana Zubatiuk, Olexandr Isayev
Accounts of Chemical Research, 2021, 54 (7), 1575–1585.
DOI: 10.1021/acs.accounts.0c00868
Machine Learned Hückel Theory: Interfacing Physics and Deep Neural Networks
Tetiana Zubatiuk, Benjamin Nebgen, Nicholas Lubbers, Justin S. Smith, Roman Zubatyuk, Guoqing Zhou, Christopher Koh, Kipton Barros, Olexandr Isayev, Sergei Tretiak
The Journal of Chemical Physics, 2021, 154 (24), 244108.
DOI: 10.1063/5.0052857
Performance and Cost Assessment of Machine Learning Interatomic Potentials
Yunxing Zuo, Chi Chen, Xiangguo Li, Zhi Deng, Yiming Chen, Joerg Behler, Gabor Csanyi, Alexander Shapeev, Aidan P. Thompson, Mitchell A. Wood, Shyue Ping Ong
Journal of Physical Chemistry A, 2020, 124 (4), 731–745.
DOI: 10.1021/acs.jpca.9b08723
Modified Embedded-Atom Method Potentials for the Plasticity and Fracture Behaviors of Unary Fcc Metals
ZH Aitken, V Sorkin, ZG Yu, S Chen, Z Wu, YW Zhang - Physical Review B, undefined 2021
APS.
Machine Learning and Computational Mathematics
E Weinan - arXiv preprint ArXiv:2009.14596, undefined 2020
arxiv.org, 1920.
Research on Microstructure and Physical Properties of Molten Carbonate Salt Based on Machine Learning
YANG Bo, L. U. Guimin
华东理工大学学报 (自然科学版), 2021, 1–11.
Machine Learning on Neutron and X-Ray Scattering and Spectroscopies
Zhantao Chen, Nina Andrejevic, Nathan C. Drucker, Thanh Nguyen, R. Patrick Xian, Tess Smidt, Yao Wang, Ralph Ernstorfer, D. Alan Tennant, Maria Chan, Mingda Li
Chemical Physics Reviews, 2021, 2 (3), 031301.
DOI: 10.1063/5.0049111
Deep Learning for Nonadiabatic Excited-State Dynamics
Wen-Kai Chen, Xiang-Yang Liu, Wei-Hai Fang, Pavlo O. Dral, Ganglong Cui
The journal of physical chemistry letters, 2018, 9 (23), 6702–6708.
DOI: 10.1021/acs.jpclett.8b03026
Building Machine Learning Force Fields of Proteins with Fragment-Based Approach and Transfer Learning
Zheng Cheng, Jiahui Du, Lei Zhang, Jing Ma, Wei Li, Shuhua Li
2021.
The Study of the Optical Phonon Frequency of 3C-SiC by Molecular Dynamics Simulations with Deep Neural Network Potential
Wei Chen, Liang-Sheng Li
Journal of Applied Physics, 2021, 129 (24), 244104.
DOI: 10.1063/5.0049464
On the Role of Gradients for Machine Learning of Molecular Energies and Forces
Anders S. Christensen, O. Anatole von Lilienfeld
Machine Learning: Science and Technology, 2020, 1 (4), 045018.
DOI: 10.1088/2632-2153/abba6f
Long-Lived Hot Electron in a Metallic Particle for Plasmonics and Catalysis: Ab Initio Nonadiabatic Molecular Dynamics with Machine Learning
Weibin Chu, Wissam A. Saidi, Oleg V. Prezhdo
ACS nano, 2020, 14 (8), 10608–10615.
DOI: 10.1021/acsnano.0c04736
Implementing a Neural Network Interatomic Model with Performance Portability for Emerging Exascale Architectures
Saaketh Desai, Samuel Temple Reeve, James F. Belak
2020.
Nonadiabatic Excited-State Dynamics with Machine Learning
Pavlo O. Dral, Mario Barbatti, Walter Thiel
The journal of physical chemistry letters, 2018, 9 (19), 5660–5663.
DOI: 10.1021/acs.jpclett.8b02469
Machine Learning and Computational Mathematics
Weinan E
2020.
Deterministic and Statistical Approaches to Quantum Chemistry
Alberto Fabrizio
2020.
The Application of Data-Driven Methods and Physics-Based Learning for Improving Battery Safety
Donal P. Finegan, Juner Zhu, Xuning Feng, Matt Keyser, Marcus Ulmefors, Wei Li, Martin Z. Bazant, Samuel J. Cooper
Joule, 2020.
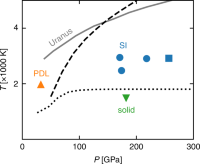
Heat and Charge Transport in H 2 O at Ice-Giant Conditions from Ab Initio Molecular Dynamics Simulations
Federico Grasselli, Lars Stixrude, Stefano Baroni
Nature communications, 2020, 11 (1), 1–7.
DOI: 10.1038/s41467-020-17275-5
Transferable Machine-Learning Model of the Electron Density
Andrea Grisafi, Alberto Fabrizio, Benjamin Meyer, David M. Wilkins, Clemence Corminboeuf, Michele Ceriotti
ACS central science, 2018, 5 (1), 57–64.
DOI: 10.1021/acscentsci.8b00551
Accuracy, Transferability, and Efficiency of Coarse-Grained Models of Molecular Liquids
M. G. Guenza, M. Dinpajooh, J. McCarty, I. Y. Lyubimov
The Journal of Physical Chemistry B, 2018, 122 (45), 10257–10278.
DOI: 10.1021/acs.jpcb.8b06687
High-Throughput Production of Force-Fields for Solid-State Electrolyte Materials
Ryo Kobayashi, Yasuhiro Miyaji, Koki Nakano, Masanobu Nakayama
APL Materials, 2020, 8 (8), 081111.
DOI: 10.1063/5.0015373
Enabling Large-Scale Condensed-Phase Hybrid Density Functional Theory Based \$ Ab \$\$ Initio \$ Molecular Dynamics II: Extensions to the Isobaric-Isoenthalpic and Isobaric-Isothermal Ensembles
Hsin-Yu Ko, Biswajit Santra, Robert A. DiStasio Jr
2020.
Molecular Dynamics Simulations of Molten Magnesium Chloride Using Machine‐Learning‐Based Deep Potential
W Liang, G Lu, J Yu
Wiley Online Library, 2020.
A Deep Neural Network Interatomic Potential for Studying Thermal Conductivity of β-Ga2O3
Ruiyang Li, Zeyu Liu, Andrew Rohskopf, Kiarash Gordiz, Asegun Henry, Eungkyu Lee, Tengfei Luo
Applied Physics Letters, 2020, 117 (15), 152102.
DOI: 10.1063/5.0025051
Effects of Density and Composition on the Properties of Amorphous Alumina: A High-Dimensional Neural Network Potential Study
Wenwen Li, Yasunobu Ando, Satoshi Watanabe
The Journal of Chemical Physics, 2020, 153 (16), 164119.
DOI: 10.1063/5.0026289
Automatically Growing Global Reactive Neural Network Potential Energy Surfaces: A Trajectory-Free Active Learning Strategy
Qidong Lin, Yaolong Zhang, Bin Zhao, Bin Jiang
2020, 152 (15).
DOI: 10.1063/5.0004944
Active Learning for Robust, High-Complexity Reactive Atomistic Simulations
Rebecca K. RK Lindsey, LE Laurence E. Fried, N Goldman - The Journal of Chemical …, undefined 2020, Nir Goldman, Sorin Bastea
2020, 153 (13).
DOI: 10.1063/5.0021965
Future Directions of Chemical Theory and Computation
Yuyuan Lu, Geng Deng, Zhigang Shuai
Pure and Applied Chemistry, 2021.
DOI: 10.1515/pac-2020-1006
A Universal Approximation Theorem of Deep Neural Networks for Expressing Probability Distributions
Yulong Lu, Jianfeng Lu
2020.
Understanding Simple Liquids through Statistical and Deep Learning Approaches
A. Moradzadeh, N. R. Aluru
The Journal of Chemical Physics, 2021, 154 (20), 204503.
DOI: 10.1063/5.0046226
Atomistic Structure Learning Algorithm with Surrogate Energy Model Relaxation
HL Henrik Lund Mortensen, Søren Ager SA Meldgaard, Malthe Kjær Bisbo, Mads Peter V. Christiansen, Bjørk Hammer, MK Bisbo - Physical Review B, undefined 2020
2020, 102 (7).
DOI: 10.1103/physrevb.102.075427
Machine Learning in Nano-Scale Biomedical Engineering
BPN Behler-Parrinello Network
.
Ring Polymer Molecular Dynamics and Active Learning of Moment Tensor Potential for Gas-Phase Barrierless Reactions: Application to S + H2
IS Ivan S. Novikov, Alexander V. Shapeev, Yury V. Suleimanov, AV Shapeev - The Journal of chemical …, undefined 2019
2019, 151 (22).
DOI: 10.1063/1.5127561
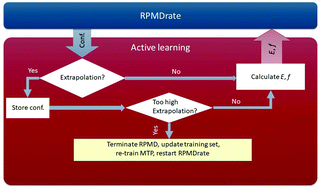
Automated Calculation of Thermal Rate Coefficients Using Ring Polymer Molecular Dynamics and Machine-Learning Interatomic Potentials with Active Learning
Ivan S. Novikov, Yury V. Suleimanov, Alexander V. Shapeev
Physical Chemistry Chemical Physics, 2018, 20 (46), 29503–29512.
DOI: 10.1039/C8CP06037A
Modeling H 2 O/Rutile-TiO 2 (110) Potential Energy Surfaces with Deep Networks
Stefan Oehmcke, Thomas Teusch, Thorben Petersen, Thorsten Klüner, Oliver Kramer
2020 International Joint Conference on Neural Networks (IJCNN), 2020, 1–7.
DOI: 10.1109/IJCNN48605.2020.9207275
Deep Learning Interatomic Potential for Simulation of Radiation Damage in Vanadium-Rich V-Cr-Ti Ternary Alloys
H. S. M. Phuong, M. D. Starostenkov, N. T. H. Trung
Эволюция Дефектных Структур в Конденсированных Средах, 2020, 141–142.
Development of a General-Purpose Machine-Learning Interatomic Potential for Aluminum by the Physically Informed Neural Network Method
GPP P.Purja Pun, V. Yamakov, J. Hickman, E. H. Glaessgen, Y. Mishin, EH Glaessgen - Physical Review …, undefined 2020
2020, 4 (11).
DOI: 10.1103/physrevmaterials.4.113807
Four Generations of High-Dimensional Neural Network Potentials
J Behler - Chemical Reviews, undefined 2021
ACS Publications.
Representing Local Atomic Environment Using Descriptors Based on Local Correlations
Amit Samanta
The Journal of chemical physics, 2018, 149 (24), 244102.
DOI: 10.1063/1.5055772
Unsupervised Learning of Atomic Environments from Simple Features
WF Reinhart - Computational Materials Science, undefined 2021
Elsevier.
A Systematic Approach to Generating Accurate Neural Network Potentials: The Case of Carbon
Y Shaidu, E Küçükbenli, R Lot, F Pellegrini
nature.com.
Elinvar Effect in β-Ti Simulated by on-the-Fly Trained Moment Tensor Potential
AV Shapeev, EV Podryabinkin, K Gubaev
iopscience.iop.org.
Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials
Andreas Singraber, Jörg Behler, Christoph Dellago
Journal of chemical theory and computation, 2019, 15 (3), 1827–1840.
DOI: 10.1021/acs.jctc.8b00770
Machine-Learned Interatomic Potentials by Active Learning: Amorphous and Liquid Hafnium Dioxide
G Sivaraman, AN Krishnamoorthy, M Baur - npj Computational …, undefined 2020
nature.com.
Automated Discovery of a Robust Interatomic Potential for Aluminum
JS Smith, B Nebgen, N Mathew, J Chen
nature.com.
Efficient Estimation of Material Property Curves and Surfaces via Active Learning
Yuan Tian, Dezhen Xue, Ruihao Yuan, Yumei Zhou, Xiangdong Ding, Jun Sun, Turab Lookman, J Sun - Physical Review …, undefined 2021
2021, 5 (1).
DOI: 10.1103/physrevmaterials.5.013802
Generalizable Protein Interface Prediction with End-to-End Learning
R. J. Townshend, Rishi Bedi, Ron O. Dror
2018.
Fast and Accurate Molecular Property Prediction: Learning Atomic Interactions and Potentials with Neural Networks
Masashi Tsubaki, Teruyasu Mizoguchi
The journal of physical chemistry letters, 2018, 9 (19), 5733–5741.
DOI: 10.1021/acs.jpclett.8b01837
Towards Modeling Spatiotemporal Processes in Metal–Organic Frameworks
Veronique Van Speybroeck, Sander Vandenhaute, Alexander EJ Hoffman, Sven MJ Rogge
Trends in Chemistry, 2021.
DOI: 10.1016/j.trechm.2021.04.003
Uncertainty Quantification in Molecular Simulations with Dropout Neural Network Potentials
M Wen, EB Tadmor
nature.com, 2020.
Deep Learning for UV Absorption Spectra with SchNarc: First Steps toward Transferability in Chemical Compound Space
Julia Westermayr, Philipp Marquetand
The Journal of Chemical Physics, 2020, 153 (15), 154112.
DOI: 10.1063/5.0021915

Machine Learning for Nonadiabatic Molecular Dynamics
Julia Westermayr, Philipp Marquetand
Machine Learning in Chemistry, 2020, 17, 76.
DOI: 10.1039/9781839160233-00076
A Data-Driven Construction of the Periodic Table of the Elements
Michael J. Willatt, Félix Musil, Michele Ceriotti
2018.
Theory and Practice of Atom-Density Representations for Machine Learning
Michael J. Willatt, Félix Musil, Michele Ceriotti
arXiv preprint, 2018.
Modeling and Predicting Responses of Magnetoelectric Materials
Ben Xu, Ce-Wen Nan
MRS Bulletin, 2018, 43 (11), 829–833.
DOI: 10.1557/mrs.2018.259
Theoretical Investigation of Halide Perovskites for Solar Cell and Optoelectronic Applications
Jingxiu Yang, Peng Zhang, Jianping Wang, Su Huai Wei
Chinese Physics B, 2020, 29 (10).
DOI: 10.1088/1674-1056/abb3f6
OnsagerNet: Learning Stable and Interpretable Dynamics Using a Generalized Onsager Principle
Haijun Yu, Xinyuan Tian, Q Li - arXiv preprint ArXiv:2009.02327, undefined 2020, Weinan E, Qianxiao Li
arxiv.org, 2020.
Exploration of Transferable and Uniformly Accurate Neural Network Interatomic Potentials Using Optimal Experimental Design
V Zaverkin, J Kästner
iopscience.iop.org, 2021.
Discovery and Design of Soft Polymeric Bio-Inspired Materials with Multiscale Simulations and Artificial Intelligence
Chenxi Zhai, Tianjiao Li, Haoyuan Shi, Jingjie Yeo
Journal of Materials Chemistry B, 2020, 8 (31), 6562–6587.
DOI: 10.1039/D0TB00896F
Inferring Micro-Bubble Dynamics with Physics-Informed Deep Learning
Hanfeng Zhai, Guohui Hu
2021.
Arrested Phase Separation in Double-Exchange Models: Machine-Learning Enabled Large-Scale Simulation
Puhan Zhang, Gia-Wei Chern
2021.
Physically Inspired Atom-Centered Symmetry Functions for the Construction of High Dimensional Neural Network Potential Energy Surfaces
Kangyu Zhang, Lichang Yin, Gang Liu
Computational Materials Science, 2021, 186, 110071.
DOI: 10.1016/j.commatsci.2020.110071
Adaptive Genetic Algorithm for Structure Prediction and Application to Magnetic Materials
Xin Zhao, Shunqing Wu, Manh Cuong Nguyen, Kai-Ming Ho, Cai-Zhuang Wang
Handbook of Materials Modeling: Applications: Current and Emerging Materials, 2020, 2757–2776.
DOI: 10.1007/978-3-319-44680-6_73