What Can Uni-Mol Do too? | Breaking the Limits of Few-shot Molecular Property Prediction
On August 19, 2024, Shuqi Lu and Zhifeng Gao from DP Technology, in collaboration with Professor Di He from Peking University, published a research article titled "Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+" in Nature Communications. This study introduces Uni-Mol+, a deep learning algorithm that innovatively utilizes neural networks to iteratively optimize initial 3D molecular conformations, enabling precise prediction of quantum chemical properties. By progressively approximating Density Functional Theory (DFT) equilibrium conformations, Uni-Mol+ significantly enhances prediction accuracy, providing a powerful tool for high-throughput screening and new material design.
1.Research Background
Uni-Mol+ is an innovative deep learning approach designed to accelerate the prediction of quantum chemical properties. Traditional methods typically rely on 1D or 2D data for direct prediction, but these approaches often lack precision since most quantum chemical properties depend on 3D equilibrium conformations optimized via electronic structure methods such as Density Functional Theory (DFT). Uni-Mol+ employs a dual-track Transformer model architecture along with a novel training strategy to optimize conformation generation and quantum chemical property prediction.
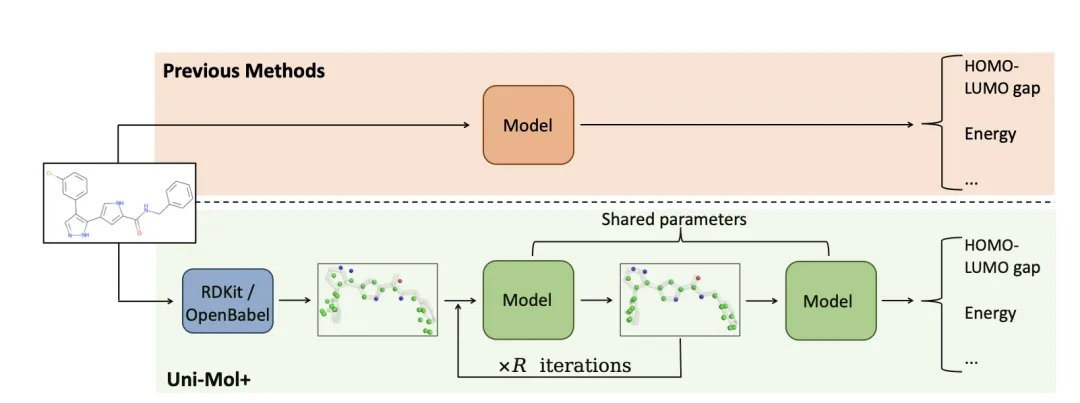
Figure 1 illustrates the Uni-Mol+ framework:
Initial 3D Conformation Generation:
Uni-Mol+ begins by generating initial 3D molecular conformations from 1D/2D data using low-cost tools such as RDKit or OpenBabel.Iterative Refinement:
The initial conformations, though generated quickly, are often not accurate enough. Uni-Mol+ iteratively refines these conformations using neural networks, progressively approximating the DFT-optimized equilibrium conformations.Final Prediction:
Once the conformation optimization is complete, Uni-Mol+ predicts quantum chemical properties based on the refined 3D conformations, significantly improving prediction accuracy.
Experimental results demonstrate that Uni-Mol+ outperforms all existing state-of-the-art methods on several public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. This highlights its immense potential for accelerating high-throughput screening, as well as the design of new materials and molecules.
2.Related Advances in the Field
1. Deep Learning Models Based on 1D/2D Information
Early studies primarily relied on 1D information (e.g., SMILES sequences) or 2D molecular graphs to predict quantum chemical properties. While these approaches are data-accessible, their predictive accuracy is often limited, as most quantum chemical properties depend on 3D equilibrium conformations optimized using Density Functional Theory (DFT).
2. Deep Learning Models Incorporating 3D Information
In recent years, there has been a growing trend toward incorporating 3D conformation information to enhance the performance of prediction models. For example, some methods integrate 3D structural data during training to improve the accuracy of 2D representations. However, during inference, these models still primarily rely on 2D information.
3. 3D Conformation Optimization and Prediction
Recent studies have focused on predicting properties directly from 3D conformations, emphasizing models' rotational and translational invariance. For instance, Uni-Mol uses 3D conformations generated by RDKit as input and introduces 3D positional recovery tasks during pretraining. However, these methods rely solely on the initial 3D conformations during inference and fail to fully optimize them to DFT-equilibrium conformations.
4. Challenges in Molecular Conformation Optimization
Molecular conformation optimization is a critical challenge in computational chemistry. While traditional DFT methods are accurate, they are computationally expensive. To improve efficiency, several deep learning-based potential energy models have been developed. These models use neural networks to replace DFT’s high-cost calculations but still require multiple iterations to optimize conformations.
In contrast, Uni-Mol+ achieves comparable results with only a few optimization iterations. It offers an end-to-end framework that simultaneously optimizes conformations and predicts quantum chemical properties, significantly improving efficiency while maintaining accuracy.
3.Methods
For any molecule, Uni-Mol+ begins by generating an initial 3D conformation using low-cost methods, such as template-based approaches from RDKit or OpenBabel. Next, it iteratively refines the initial conformation toward the target conformation, which is the DFT-optimized equilibrium conformation. Finally, based on the refined conformation, Uni-Mol+ predicts quantum chemical properties.
To achieve this, a novel model architecture and a new training strategy were introduced to optimize conformations and predict quantum chemical properties. These aspects are discussed in detail in the following sections.
3.1 Model Architecture
The research team designed a novel model architecture capable of simultaneously learning molecular conformations and predicting quantum chemical properties, represented as ( (y, \hat{r}) = f(X, E, r; \theta) ). This model uses atomic features ((X \in \mathbb{R}^{n \times d_f}), where (n) is the number of atoms and (d_f) is the dimension of atomic features), edge features ((E \in \mathbb{R}^{n \times n \times d_e}), where (d_e) is the dimension of edge features), and atomic 3D coordinates ((r \in \mathbb{R}^{n \times 3})). By optimizing the parameters (\theta), it predicts quantum chemical properties (y) and updates the 3D coordinates (\hat{r} \in \mathbb{R}^{n \times 3}).
This architecture preserves two representation pathways similar to Uni-Mol:
- Atomic representation ((x \in \mathbb{R}^{n \times d_x}), where (d_x) is the atomic feature dimension).
- Pairwise representation ((p \in \mathbb{R}^{n \times n \times d_p}), where (d_p) is the pairwise feature dimension).
The model consists of a single block, with (x^{(l)}) and (p^{(l)}) as the outputs of the (l)-th block.
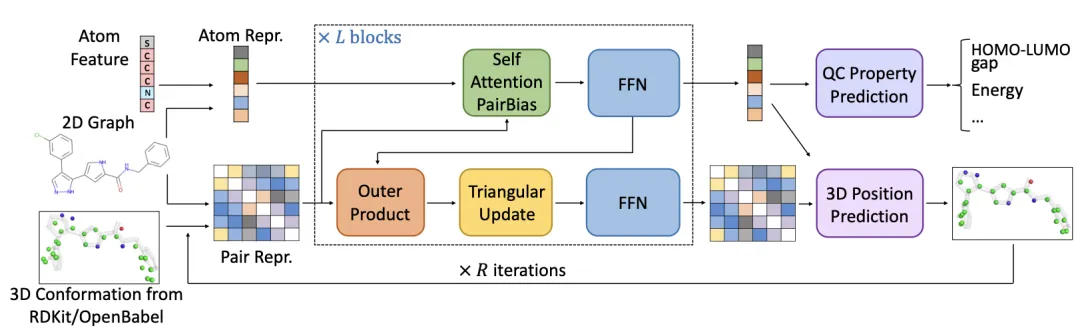
Figure 2 illustrates the overall architecture. Compared to Uni-Mol, there are several key differences:
Position Encoding:
The model uses positional encoding methods to embed 3D spatial and 2D graph positional information. Specifically, for 3D spatial information, Gaussian kernels are used for encoding, similar to previous research. Additionally, graph positional encodings, such as shortest-path and hop encodings, are introduced.Atomic Representation Update:
In each block, atomic representations are iteratively updated through an attention mechanism and feed-forward neural networks.Pairwise Representation Update:
Pairwise representations are updated through outer products, triangle updates, and feed-forward neural networks, further enhancing the expressiveness of pairwise representations.
3.2 Model Training
In DFT conformation optimization or molecular dynamics simulations, conformations are iteratively optimized, forming a trajectory from the initial conformation to the equilibrium conformation. However, saving such trajectories can be very costly, and public datasets typically provide only equilibrium conformations. To fully leverage this information, we propose a novel training method that first generates a pseudo-trajectory, samples a conformation from it as input, and uses it to predict the equilibrium conformation.
4.Experimental Validation of Uni-Mol+'s Exceptional Performance
In this study, the research team conducted a thorough empirical analysis of the Uni-Mol+ model’s performance. The team first detailed the model's configuration and then performed comprehensive benchmarking using two widely recognized public datasets, PCQM4MV2 and OC20, to evaluate the model’s capabilities in small organic molecules and catalytic systems. Finally, ablation studies were conducted to analyze the impact of different model components and training strategies on overall performance, further confirming the superiority of Uni-Mol+.
4.1 Model Configuration
The Uni-Mol+ model adopts a 12-layer architecture, with the following configurations:
- Atom representation: Dimension = 768
- Pair representation: Dimension = 256
- FFN hidden layer dimensions:
- Atom representation track: 768
- Pair representation track: 256
- OuterProduct hidden dimension: 32
- TriangularUpdate hidden dimension: 32
The model performs a single conformation optimization iteration, indicating a total of two iterations: one for conformation optimization and one for quantum chemical property prediction.
Training strategy:
- Standard deviation of random noise: 0.2
- Sampling of parameter ( q ):
- ( q = 0.0 ) with probability 0.8
- ( q = 1.0 ) with probability 0.1
- Uniform sampling from [0.4, 0.6] with probability 0.1
- Total model parameters: approximately 52.4M
4.2 PCQM4MV2 Dataset
Background:
The PCQM4MV2 dataset originates from the OGB Large-Scale Challenge and aims to advance and evaluate machine learning models for molecular quantum chemical property prediction, particularly for the HOMO-LUMO gap, which represents the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The dataset consists of approximately 4 million molecules represented using SMILES. While the training and validation sets include HOMO-LUMO gap labels, the test set labels remain undisclosed. The training set also provides DFT-optimized equilibrium conformations, but the validation and test sets do not. The benchmarking objective is to predict the HOMO-LUMO gap using SMILES during inference, without relying on DFT equilibrium conformations.
Experimental Setup:
- Initial conformations: 8 initial conformations per molecule were generated using RDKit (approx. 0.01 seconds per molecule).
- Training: During each epoch, one conformation was randomly sampled as input.
- Inference: The average HOMO-LUMO gap prediction over 8 conformations was used.
Hyperparameters:
- Optimizer: AdamW
- Learning rate: ( 2 \times 10^{-4} )
- Batch size: 1024
- ( \beta_1 = 0.9 ), ( \beta_2 = 0.999 )
- Gradient clipping: 5.0
- Training duration: 1.5 million steps (including 150k warm-up steps)
- Exponential Moving Average (EMA) decay rate: 0.999
Training Infrastructure:
- Training: 8 NVIDIA A100 GPUs, approximately 5 days.
- Inference: 8 NVIDIA V100 GPUs, approximately 7 minutes for the 147k test-dev set.
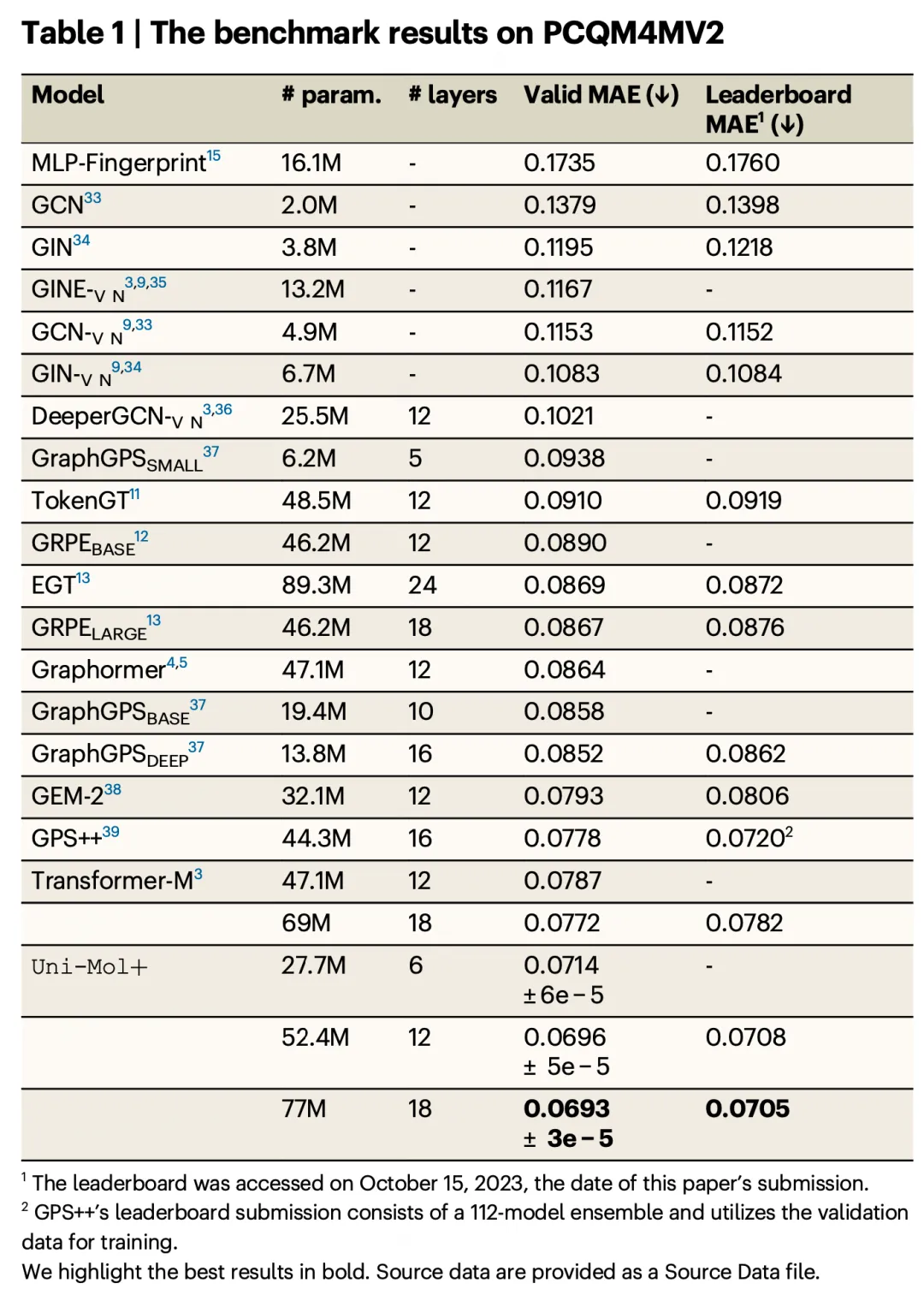
Performance Results on PCQM4MV2
The team compared the previous leaderboard submissions on PCQM4MV2 as baselines and evaluated three variants of Uni-Mol+ models (6-layer, 12-layer, and 18-layer) to study the impact of model size on performance.
Results Summary (Table 1):
- All three Uni-Mol+ variants achieved significant performance improvements over previous baseline methods.
- Even the 6-layer Uni-Mol+ model, despite having fewer parameters, outperformed all prior baseline methods.
- Increasing the number of layers from 6 to 12 resulted in a substantial accuracy improvement, far exceeding all baseline methods.
- The 18-layer Uni-Mol+ model delivered the best performance, achieving a significant edge over all baselines.
4.3 Open Catalyst 2020
The Open Catalyst 2020 (OC20) dataset is designed to advance machine learning models for catalyst discovery and optimization. OC20 includes three tasks: Structure to Energy and Forces (S2EF), Initial Structure to Relaxed Structure (IS2RS), and Initial Structure to Relaxed Energy (IS2RE). This study focuses on the IS2RE task, which aligns with the objectives of the proposed method. The IS2RE task aims to predict the relaxed energy based on the initial conformation. The dataset contains approximately 460,000 training data points. While DFT-equilibrium conformations are available during training, their use is prohibited during inference. Unlike the PCQM4MV2 dataset, the initial conformations are already provided in OC20, eliminating the need to generate initial input conformations.
Experimental Setup
For the OC20 experiments, the team used the default 12-layer Uni-Mol+ configuration with some modifications compared to the PCQM4MV2 setup:
Exclusion of Graph Features:
Since the OC20 dataset lacks graph information, all graph-related features were excluded from the model.Model Capacity Adjustment:
Due to the larger number of atoms in the OC20 dataset compared to PCQM4MV2, the model capacity was slightly reduced for efficiency. Specifically:- Pair representation dimension: 128
- OuterProduct hidden dimension: 16
- TriangularUpdate hidden dimension: 16
Periodic Boundary Conditions:
To account for periodic boundary conditions, the team employed a pre-expansion of neighboring cells combined with a radius cutoff to reduce the number of atoms considered.Training Configuration:
- Optimizer: AdamW
- Training steps: 1.5 million (including 150,000 warm-up steps)
- Learning rate: (2 \times 10^{-4})
- Batch size: 64
- ( \beta_1 = 0.9 ), ( \beta_2 = 0.999 )
- Gradient clipping: 5.0
The training process took approximately 7 days using 16 NVIDIA A100 GPUs.
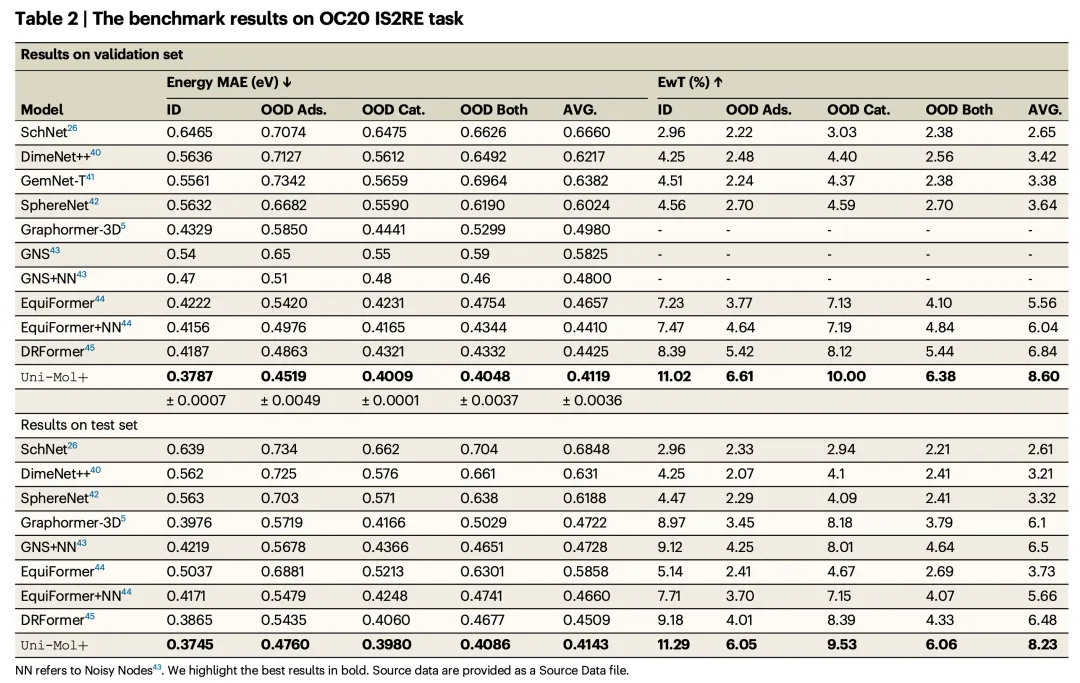
Results
Tables 2 and 3 present the performance comparisons of various models on the OC20 IS2RE validation and test sets. Metrics include:
- Mean Absolute Error (MAE) for energy predictions
- Energy within Threshold (EwT%), which measures the percentage of energy predictions falling within a certain threshold.
Key Findings:
- Significant Improvements: Uni-Mol+ outperformed all prior baseline methods in both MAE and EwT across all categories.
- Lowest MAE Across Categories:
- Uni-Mol+ achieved the lowest MAE values across all categories, including:
- In-domain (ID)
- Out-of-domain Adsorption (OOD Ads.)
- Out-of-domain Catalyst (OOD Cat.)
- Out-of-domain Both (OOD Both)
- Average (AVG)
- Uni-Mol+ achieved the lowest MAE values across all categories, including:
- Highest EwT Scores:
- Uni-Mol+ also achieved the highest EwT scores in all categories, emphasizing its robustness across both in-domain and out-of-domain data.
Conclusions
In this study, the Uni-Mol+ model introduces a novel approach to quantum chemical property prediction using deep learning. Unlike traditional methods, Uni-Mol+ begins by leveraging low-cost tools to generate initial 3D conformations from 1D/2D data. These conformations are then iteratively optimized to DFT-equilibrium conformations through neural networks, and quantum chemical properties are predicted based on the optimized conformations.
The results demonstrate that Uni-Mol+ significantly outperforms all previous methods across multiple public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. Ablation studies further validate the effectiveness of each component of the model.
Overall, Uni-Mol+ showcases immense potential for accelerating quantum chemical property prediction through deep learning, providing an effective tool for high-throughput screening, new material discovery, and novel molecule design.