What Can DP Do too? | Machine Learning Interatomic Potential for Mg-Al-Si-O System Boosts Deep Earth Material Research
Phase diagrams and thermodynamic properties of deep Earth materials are fundamental to geophysics, geodynamics and geological research. As the core chemical system of mantle mineralogy, the Mg-Al-Si-O system governs the stability and physical properties of dominant mantle minerals and melts, further modulating mantle dynamics and plate tectonics. Recently, Xin Zhong, Timm John from Free University of Berlin and Yifan Li from Princeton University published their research A general purposed machine learning interatomic potential for Mg-Al-Si-O system suitable for Earth materials at high pressure and temperature conditions in npj Computational Materials. The team developed a universal machine learning interatomic potential for the Mg-Al-Si-O system. By combining the r2SCAN functional with pairwise Gaussian energy correction, the average enthalpy error of over 20 mineral phases was reduced from 5.2 kJ/mol to 1.2 kJ/mol. The potential reproduces phase diagrams of systems including SiO2 Al2SiO5 and Mg2SiO4 with excellent consistency against experimental measurements. The study quantitatively characterizes the anisotropy of solid–melt interfacial free energy for periclase and forsterite at the atomic scale, and quantifies how non-hydrostatic stress modulates the α-β quartz phase transition.
Research Background
The Mg-Al-Si-O chemical system underpins mantle mineralogy, encompassing major mineral endmembers such as olivine, pyroxene, spinel, garnet and bridgmanite. High-pressure mineral phase transitions account for seismic discontinuities within the Earth’s interior, while accurate phase diagrams rely on precise thermodynamic property data. Although laboratory experiments constrain these properties, atomistic simulations — including molecular simulations with interatomic potentials and density functional theory (DFT) calculations — have emerged as powerful tools for investigating mineral elasticity, surface energy, atomic diffusion and other critical properties. Nevertheless, conventional empirical potentials face severe limitations when modeling large-scale systems and complex phenomena such as phase transitions. Machine learning interatomic potentials trained on high-fidelity DFT datasets, especially those constructed with meta-GGA functionals like SCAN, exhibit exceptional capability to reproduce experimental phase diagrams and thermodynamic observables.
High-Precision Deep Potential Model: A Novel Tool for Earth Material Simulations
This work develops a machine learning interatomic potential for the Mg-Al-Si-O system based on the DeePMD-kit framework. The training dataset covers more than 20 typical rock-forming minerals and their corresponding melts. Initial atomic configurations are primarily sampled from Car–Parrinello molecular dynamics (CPMD) simulations spanning 300–3000 K and 0–50 GPa, supplemented by random structural perturbations of known crystal structures. An active learning strategy analogous to the DP-GEN workflow is adopted to iteratively enhance model robustness and applicable domain via a closed loop: potential training → novel configuration generation → training set augmentation.
The Al2SiO5 polymorph system (kyanite–andalusite–sillimanite) is selected as a benchmark to benchmark the performance of exchange-correlation functionals. This system features tiny energy gaps between distinct mineral phases, rendering it highly sensitive to functional accuracy and thus ideal for validating thermodynamic calculations of geologic materials. Results demonstrate that the r2SCAN functional yields energy errors below 3 kJ/mol for this system, alongside stable self-consistent field convergence, making it well-suited for subsequent melt simulations and training dataset construction.
A core technical advance, pairwise Gaussian energy correction, is implemented to further refine phase stability predictions. This correction scheme introduces subtle adjustments to inter-element interaction energies while preserving the intrinsic accuracy and atomic interaction features of r2SCAN, aligning enthalpy values computed from the machine learning potential with reference thermodynamic databases.
The correction drastically improves the model’s predictive power for mineral phase stability. For over 20 representative mineral phases, the average enthalpy error decreases from 5.2 kJ/mol to 1.2 kJ/mol, with negligible degradation in volume prediction accuracy. This confirms the model retains reliable structural descriptions while achieving drastically improved thermodynamic precision.
(See Figure 1 for the full potential construction workflow; Figure 2 for the performance of Gaussian energy correction.)
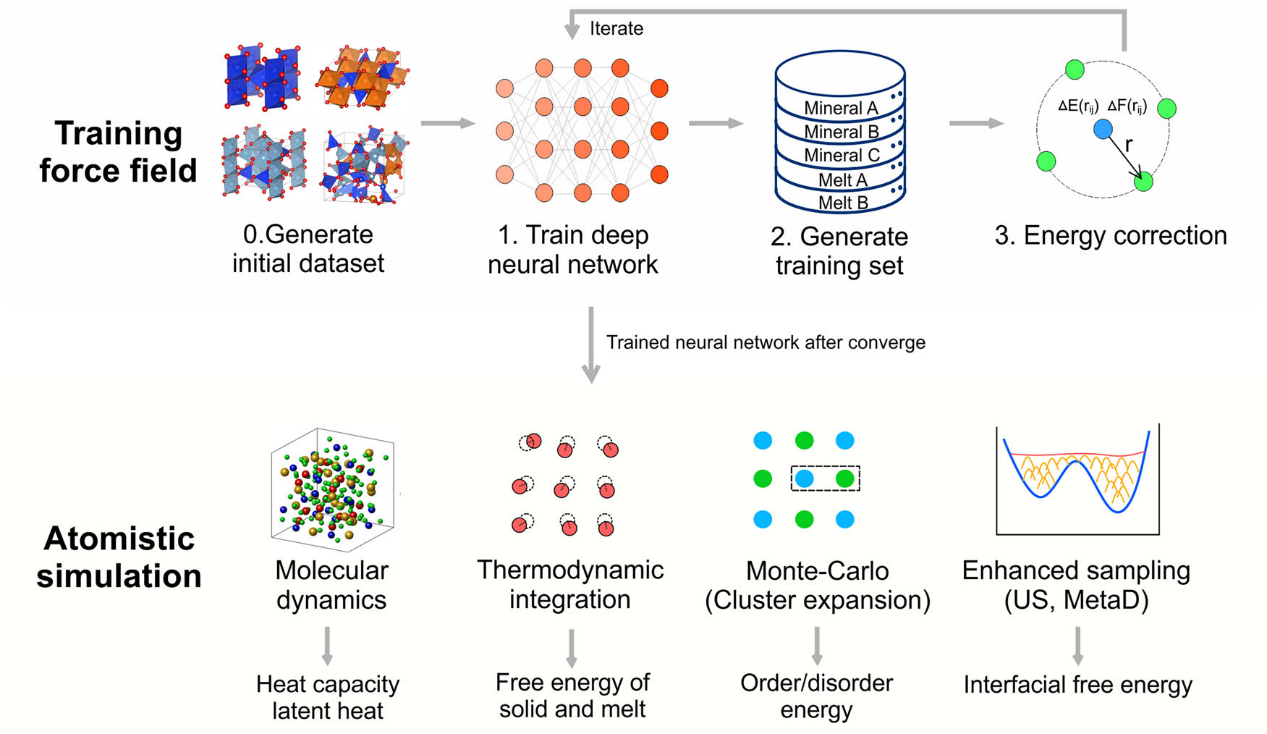
Figure 1 Workflow of machine learning potential development and application. The upper panel illustrates initial dataset generation, neural network training, active learning dataset expansion and Gaussian energy correction. The lower panel lists downstream applications enabled by this potential, including molecular dynamics, thermodynamic integration, Monte Carlo/cluster expansion and enhanced sampling simulations.
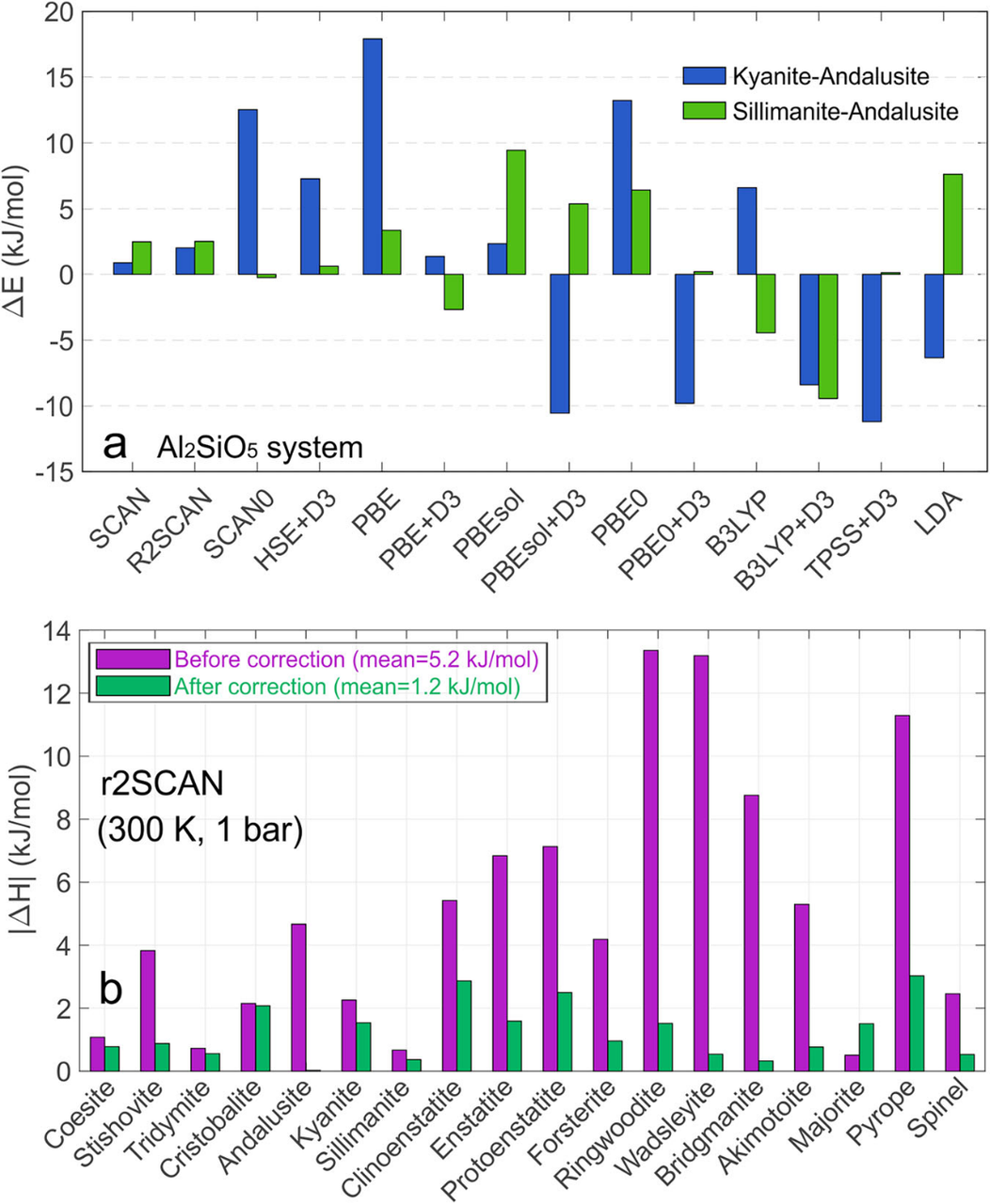
Figure 2 Functional benchmarking and performance of Gaussian energy correction. The upper subplot compares relative energy errors of different functionals for the Al2SiO5 system. The lower subplot shows marked reductions in enthalpy errors for all mineral phases after correction, with the average error lowered from ~5.2 kJ/mol to ~1.2 kJ/mol.
Phase Diagram Construction: Validation of Model Predictive Capacity
Equipped with the trained machine learning potential and the DPTI package developed by the DeepModeling community, the authors constructed phase diagrams for SiO2 , Al2SiO5 and Mg2SiO4. Gibbs free energies are evaluated via thermodynamic integration, and phase boundaries are located using the Clausius–Clapeyron relation, yielding predictions consistent with experimental constraints. (See Figure 3.)
For the SiO2 system, machine learning potential molecular dynamics predicts the stishovite-to-CaCl₂-type post-stishovite transition at ~53 GPa, close to the experimental value of ~60 GPa, verifying the model’s extrapolation capability. The andalusite–sillimanite phase boundary for Al2SiO5 is significantly refined after Gaussian energy correction; Monte Carlo simulations accounting for Al–Si order–disorder effects within sillimanite resolve discrepancies in P–T boundary slopes. For the Mg2SiO4 system, the potential successfully reproduces key features including forsterite melting and the bridgmanite–periclase liquidus. The authors also discuss model limitations: for instance, the ~7 GPa deviation in ultrahigh-pressure SiO2 corresponds to a depth offset of ~200 km, requiring caution when interpreting geophysical observations.
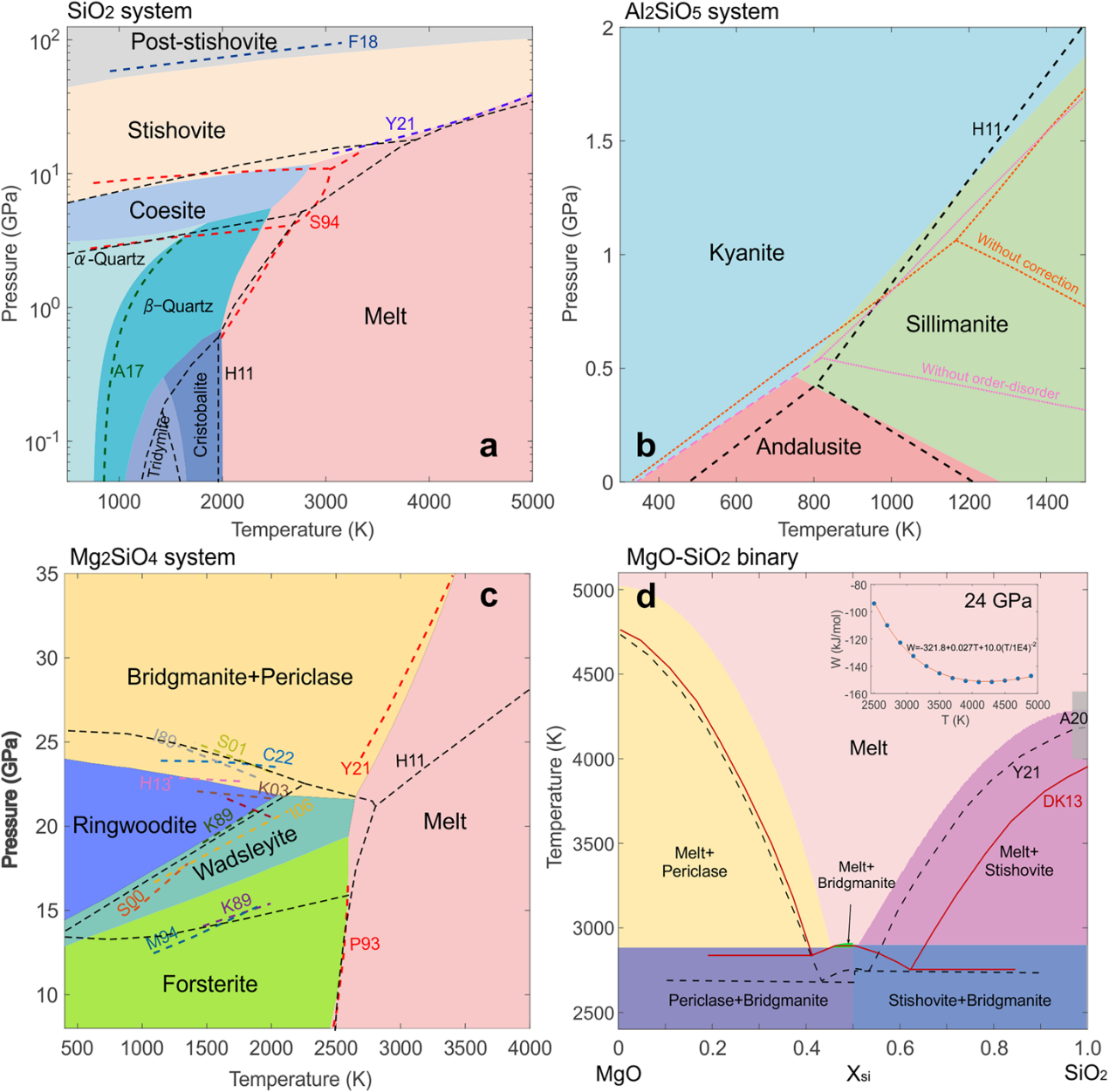
Figure 3 Phase diagrams computed using the machine learning potential. Colored regions denote model predictions, while dashed lines represent experimental or published thermodynamic constraints. The figure presents P-T phase diagrams for SiO2 , Al2SiO5 and Mg2SiO4, alongside the T-X phase diagram of the binary MgO-SiO2 system at 24 GPa.
Order–Disorder Transitions: Atomistic Insight into Sillimanite Thermodynamics
A critical feature of the Al2SiO5 system is Al-Si order–disorder within sillimanite. At elevated temperatures, Al and Si cations rearrange along chain-like structural units, modifying mixing free energy and shifting phase boundary slopes.
The machine learning potential enables efficient energy evaluation for diverse Al-Si atomic arrangements, whose mixing free energies are subsequently quantified via cluster expansion and Monte Carlo simulations. Long-range ordering rapidly decays above ~1300 K, whereas short-range ordering diminishes more gradually, indicating localized intra-chain Al-O-Si coordination persists at higher temperatures. Compared with the conventional Bragg-Williams approximation, this atomistic framework predicts mixing free energies ~25% lower, delivering refined atomic-scale constraints on sillimanite stability.
Solid–Melt Interfacial Free Energy: A Hard-to-Measure Experimental Observable
Beyond phase diagram modeling, the study calculates solid–melt interfacial free energy, a parameter inaccessible to routine laboratory characterization. This quantity governs crystal morphology, melting mechanisms, melt distribution along grain boundaries, and microstructural evolution of high-temperature mantle materials.
The computational workflow is summarized as follows: periodic supercells with coexisting solid and molten domains are constructed, and free energy profiles are obtained via umbrella sampling and metadynamics. At the melting point, the Gibbs free energy difference between a pure solid cell and a solid–melt coexistence cell arises from two solid–melt interfaces formed under periodic boundary conditions. Dividing this free energy difference by twice the interfacial area yields the solid–melt interfacial free energy.
For periclase at 24 GPa and 5200 K, metadynamics and umbrella sampling deliver consistent interfacial free energy values ranging from 0.62 to 0.66 J/m², with an interfacial anisotropy of ~ 6%. For structurally complex forsterite, metadynamics exhibits slow convergence, so umbrella sampling with chemical potential correction is adopted. Results show the (010) plane bears the lowest interfacial free energy (~ 0.29 J/m²), while the (001) plane exhibits the highest value (~ 0.33 J/m²), corresponding to an anisotropy of ~12%. Such anisotropic information cannot be directly resolved by existing experimental techniques.
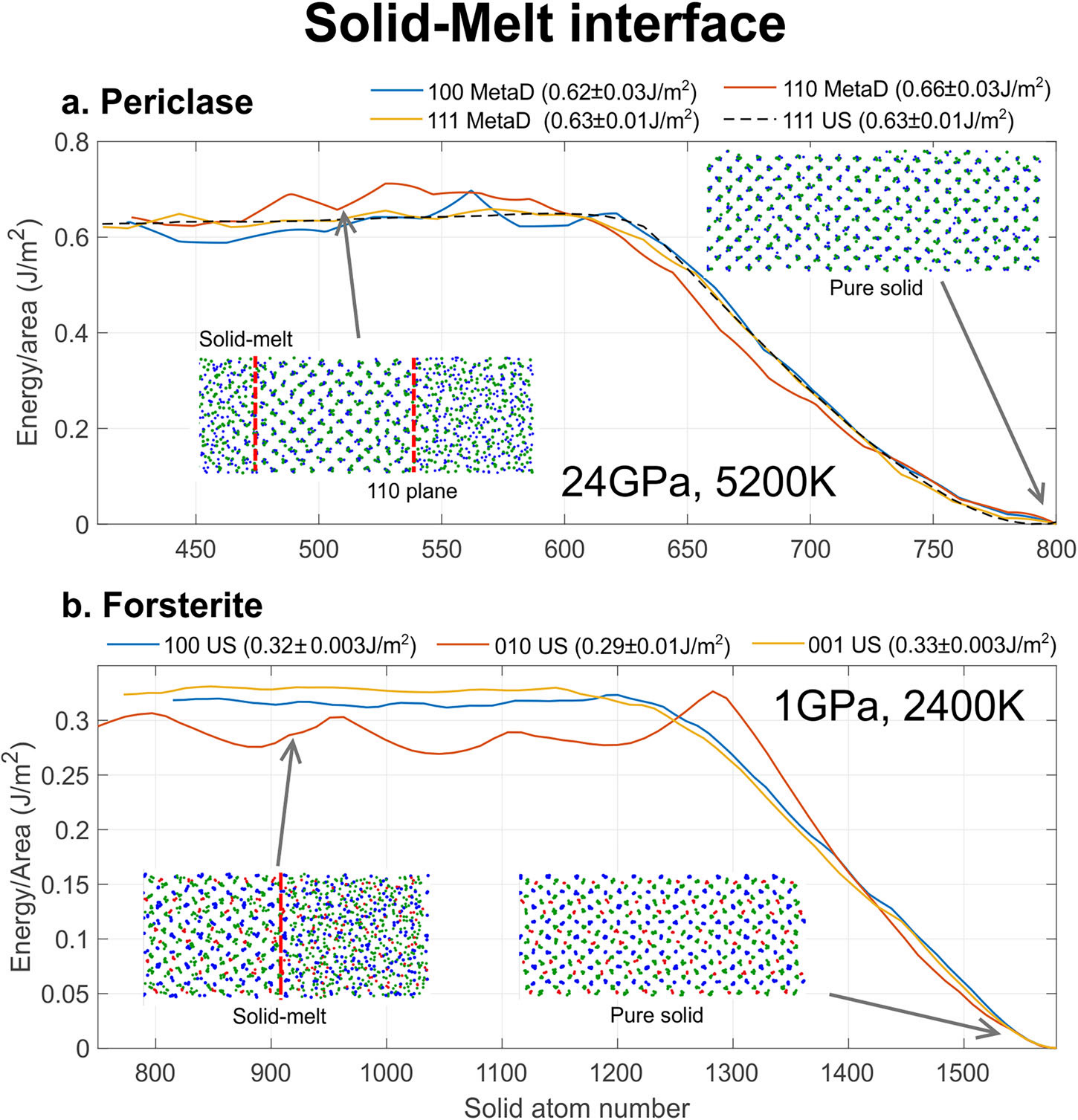
Figure 4 Calculation of solid–melt interfacial free energy. The upper subplot presents periclase–melt interfaces, showing consistent outputs from metadynamics (MetaD) and umbrella sampling (US). The lower subplot displays forsterite–melt interfacial free energies for distinct crystallographic planes, revealing ~12% anisotropy.
Non-Hydrostatic Stress Effects: New Perspective on the α-β Quartz Transition
The α-β quartz transition, driven by rotation of Si-O-Si bond angles, serves as an ideal model system to probe non-hydrostatic stress effects. Simulations at 1200 K with variable differential stress track Si-O-Si bond angle evolution as a function of mean stress. The results indicate that each 1 GPa increment in differential stress shifts the mean transition stress by ~0.17 GPa, introducing up to 17% systematic error if mean stress alone is used as the transition criterion.
Under typical crustal conditions, differential stress within quartzite commonly ranges from 0.1 to 0.3 GPa, inducing a transition pressure offset of merely 0.02–0.06 GPa, equivalent to negligible depth shifts. Therefore, non-hydrostatic stress will not drastically alter the P–T stability window of the α-β quartz transition unless extreme localized differential stress occurs.
Conclusions and Outlook
This research delivers more than a standalone Deep Potential interatomic potential; it establishes a complete, standardized workflow for computational modeling of Earth materials: appropriate DFT functionals guarantee accurate structural and energetic descriptions, active learning expands the sampled configuration space, thermodynamic database-based energy correction refines phase stability predictions, and cross-validation against experimental phase diagrams validates overall model reliability.
The resulting machine learning potential enables investigations intractable for conventional experiments or ab initio calculations, including high-P/T solid–melt interfacial free energies, order–disorder mixing free energies, stress-modulated phase transition pathways, and future simulations of complex multi-component mantle lithologies. Although the current potential only covers the four-component Mg-Al-Si-O system, this work lays a solid foundation for extensions toward multi-element solid solutions and compositional models representative of natural rock assemblages.