What Can DP Do too? | Using Machine Learning to Accelerate Molecular Dynamics to Uncover the Role of Water-Mediated Proton Hopping Mechanism at the SnO₂(110)/Water Interface
Recently, Dr. Mei Jia from Shangqiu Normal University, Dr. Yongbin Zhuang from École Polytechnique Fédérale de Lausanne (EPFL), and Prof. Jun Cheng from Xiamen University conducted an in-depth study on the proton transfer mechanism at the SnO₂(110)/H₂O interface by combining ab initio molecular dynamics (AIMD) with the Deep Potential (DP) method. The team used AIMD to obtain the electronic structure of the interface system and applied the Deep Potential Molecular Dynamics (DPMD) model to accelerate molecular dynamics simulations, enabling larger-scale and longer-timescale simulations. This combination of methods allowed the researchers to analyze the free energy distributions of different proton transfer pathways in detail and to reveal the influence of the solvation environment on the proton transfer process.
The related findings have been published in the high-impact journal Precision Chemistry, under the title “Water-Mediated Proton Hopping Mechanisms at the SnO₂(110)/H₂O Interface from Ab Initio Deep Potential Molecular Dynamics.” Dr. Mei Jia and Dr. Yongbin Zhuang are the co-first authors, and Prof. Jun Cheng is the corresponding author.
Research Background
Proton transfer (PT) reactions at metal oxide/water interfaces are key steps in various chemical processes, including photocatalytic water splitting, dehydrogenation, and hydrogen storage. In recent years, experimental characterization and theoretical simulations of PT processes have garnered significant attention. While experimental techniques can reveal the composition, structure, and certain dynamic properties of interfaces, challenges remain in detecting and identifying dynamic proton hopping phenomena at interfaces and distinguishing different PT reaction pathways.
Ab initio molecular dynamics (AIMD) simulations can accurately describe the breaking and formation of O–H bonds in the interfacial solvent environment at the atomic scale, making them suitable for studying interfacial PT reactions. However, AIMD is computationally expensive, and achieving equilibrium for PT reactions between interfacial species—especially slower processes—on the picosecond timescale is challenging.
In recent years, machine learning techniques have helped overcome the time-scale limitations of AIMD simulations, enabling in-depth studies of structural characteristics, dynamic behaviors, hydrogen-bond networks, and PT pathways at metal oxide interfaces, such as ZnO, IrO₂, CeO₂, and TiO₂. Nevertheless, significant uncertainties remain regarding the energy barriers of different PT pathways and the effects of solvation on rutile oxide surfaces.
At the SnO₂ interface, the nearly equivalent acidity constants lead to frequent proton transfers. Three distinct PT pathways have been identified at this interface (Figure 1):
Direct transfer of a proton from a terminally adsorbed water molecule to a bridge oxygen site (surface-PT),
Transfer mediated by solvent water molecules (mediated-PT), and
Transfer between two terminally adsorbed water molecules (adlayer-PT).
Thus, to explore the relationships between the microscopic structure, PT reaction pathways, and solvation effects on rutile oxide surfaces, SnO₂ was chosen as an ideal model system.

Methods
In this study, the research team employed various advanced simulation methods to thoroughly investigate the proton transfer (PT) mechanisms, as well as the microscopic structure and dynamic characteristics, at the SnO₂(110)/H₂O interface.
First, the researchers used ab initio molecular dynamics (AIMD) simulations. AIMD provides accurate descriptions of the electronic structure and the breaking and formation of O−H bonds at the atomic scale, offering foundational data for studying PT reactions at the interface. However, due to the high computational cost and time-scale limitations of AIMD, it is unsuitable for large-scale or long-duration simulations. To address this limitation, the team introduced the Deep Potential (DP) method. The DP model was trained using a large set of atomic configurations extracted from AIMD simulations, for which the energy and forces were computed. With this data, a deep neural network was trained to learn and accurately reproduce the potential energy surface of the system. This machine-learning-based model retains the accuracy of AIMD while significantly improving computational efficiency, enabling larger-scale and longer-duration simulations.
Using the trained DP model, the team further conducted Deep Potential Molecular Dynamics (DPMD) simulations. DPMD combines the efficiency of deep learning with the precision of molecular dynamics, allowing the researchers to explore the dynamic behavior and energy changes associated with different PT pathways at the SnO₂(110)/H₂O interface over longer time scales. Additionally, the study applied the Climbing Image Nudged Elastic Band (CI-NEB) method to calculate the transition states and energy barriers of the PT processes. CI-NEB optimizes images along the reaction pathway to identify the transition states, helping the researchers determine the energy barriers and reaction mechanisms of various PT pathways. This information is crucial for understanding how protons move at the interface and how they are influenced by the solvation environment.
Results and Discussion
DPMD Simulations Reconstruct Key Structures and Dynamic Behaviors
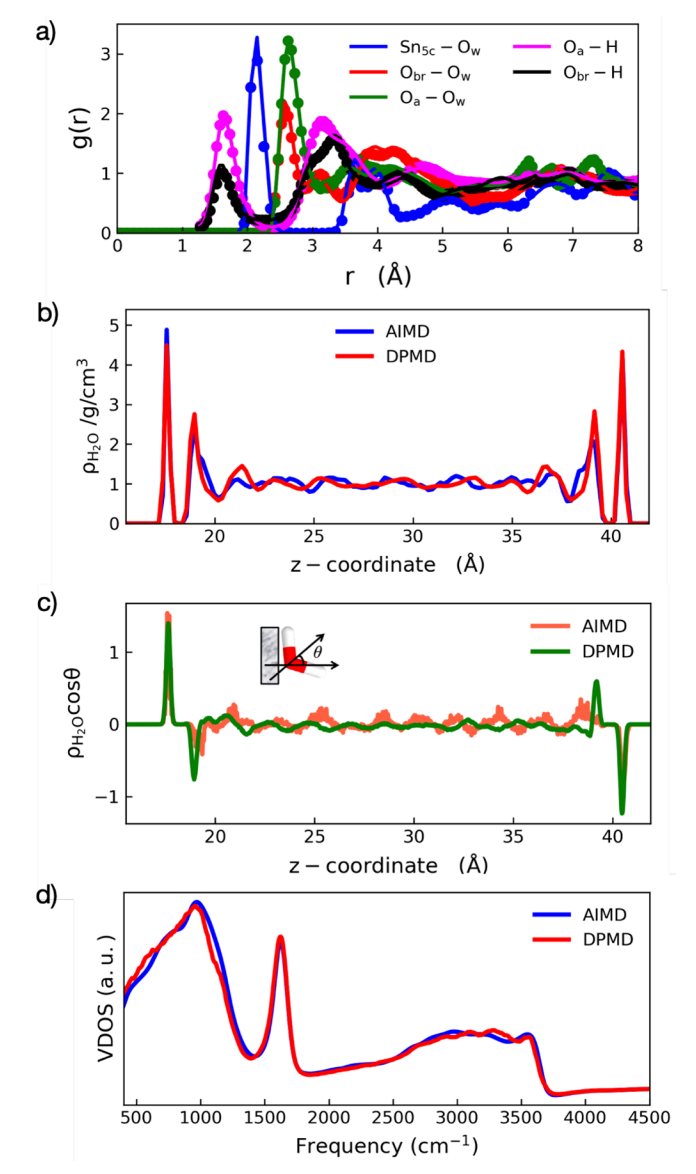
Figure 2 illustrates the key structures and dynamic characteristics of the SnO₂(110)/H₂O interface obtained from Deep Potential Molecular Dynamics (DPMD) and ab initio molecular dynamics (AIMD) simulations. The figure includes the radial distribution function (RDF) for selective atomic pairs, the density distribution of interfacial water, the dipole orientation of water molecules, and the vibrational density of states (VDOS) for interfacial O-H groups. A comparison between DPMD and AIMD results reveals consistency in predicting the structure and behavior of interfacial water molecules. These analyses demonstrate that DPMD simulations can accurately capture the arrangement of water molecules, hydrogen-bond networks, and vibrational characteristics at the SnO₂(110)/H₂O interface, providing a solid foundation for subsequent in-depth studies of proton transfer mechanisms.
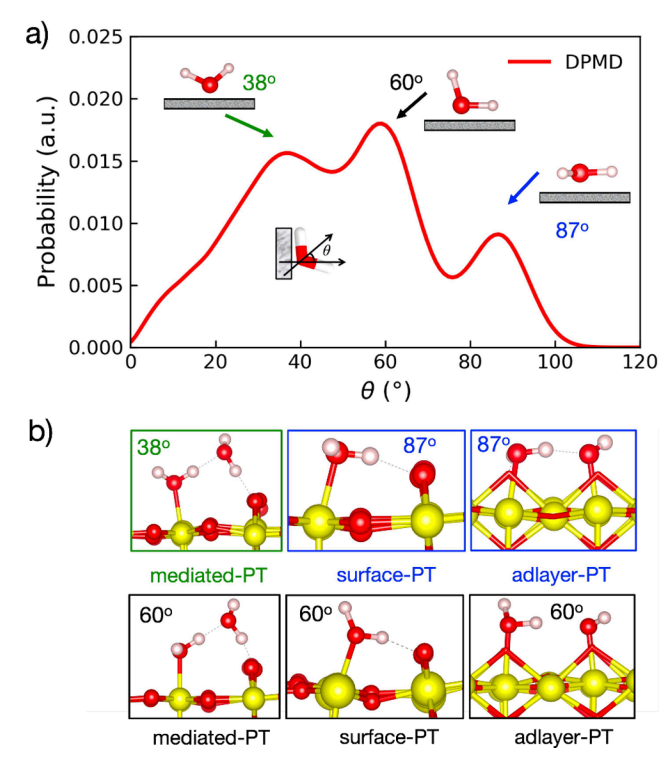
Orientation Distribution of Interfacial Water Molecules Reveals Proton Transfer Mechanisms
Figure 3 shows the orientation distribution of terminally adsorbed water molecules at the SnO₂(110)/H₂O interface and their relationship with proton transfer (PT) pathways. Figure 3a presents the probability distribution of the angle between the dipole moment of terminal water molecules and the surface normal, revealing three main peaks corresponding to different water orientations. Figure 3b uses these orientations to detail the three types of PT mechanisms: direct PT from terminal water to bridge oxygen (surface-PT), indirect PT mediated by solvent water (mediated-PT), and PT between two terminally adsorbed water molecules (adlayer-PT). By analyzing the relationship between water molecule orientations and PT pathways, researchers were able to uncover the significant influence of the interfacial solvation environment on the proton transfer process.
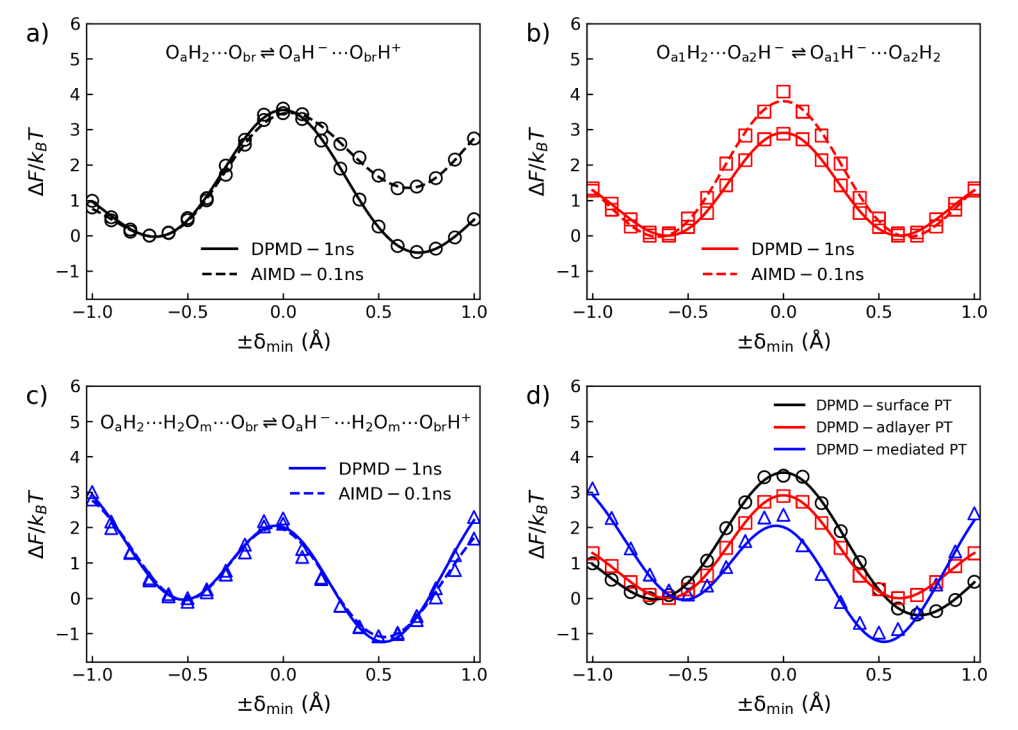
Free Energy Curves Reveal Proton Transfer Pathways at the SnO₂(110)/H₂O Interface
Figure 4 displays the free energy distributions of three proton transfer (PT) pathways at the SnO₂(110)/H₂O interface, obtained through AIMD and DPMD simulations. Figures 4a to 4c correspond to surface-PT, adlayer-PT, and mediated-PT, respectively. Figure 4d compares the free energy curves of the three PT pathways, showing that mediated-PT has the lowest energy barrier and the fastest relative reaction rate. These free energy analyses reveal the thermodynamic and kinetic characteristics of different PT pathways at the interface, providing critical insights into the influence of the solvation environment on the proton transfer process.
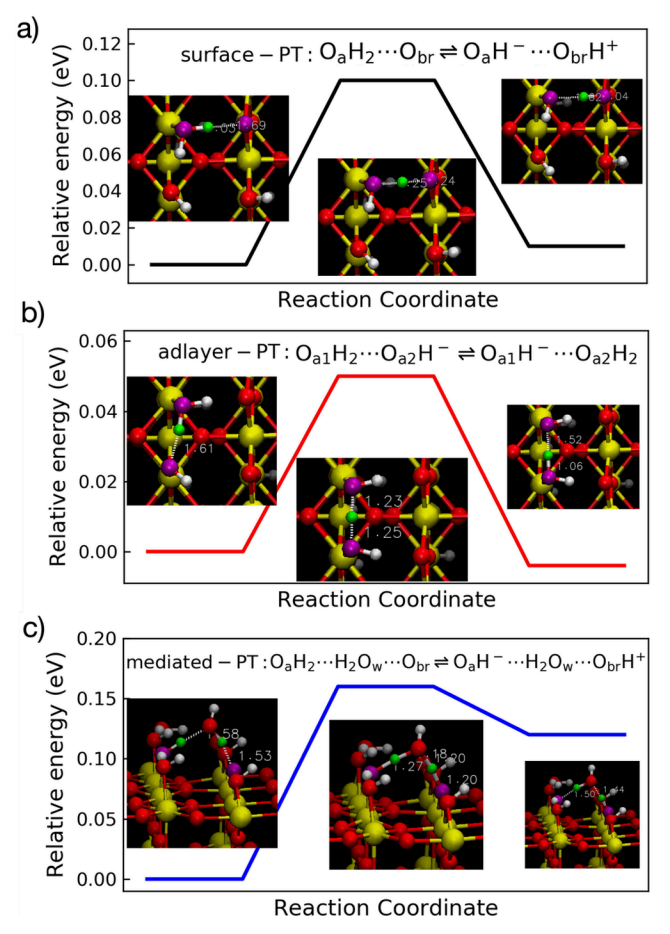
Minimum Energy Path Analysis for the SnO₂(110) Surface
Figure 5 presents the minimum energy paths (MEPs) of three PT pathways on the SnO₂(110) surface, calculated using the Climbing Image Nudged Elastic Band (CI-NEB) method. The pathways include surface-PT, adlayer-PT, and mediated-PT. By optimizing the images along the reaction path, the CI-NEB method identifies the transition states and energy barriers for the reactions. The results in Figure 5 indicate that mediated-PT has the highest energy barrier, while surface-PT and adlayer-PT have relatively lower energy barriers. These calculations provide theoretical support for understanding how protons transfer through different pathways on the SnO₂(110) surface.
Proton Transfer Mechanisms on Metal Oxide Interfaces
Figure 6 summarizes schematic diagrams of proton transfer (PT) mechanisms on metal oxide surfaces as reported in different studies. The figure illustrates seven types of PT pathways, including direct PT from terminal water to surface oxygen, mediated PT via solvent water, direct and mediated PT between surface oxygen atoms, and direct or multi-mediated PT between terminal groups. These diagrams help elucidate proton transfer behaviors on various oxide surfaces in solution environments, highlighting the roles of proton donors and acceptors, hydrogen bonds, and solvation effects during PT processes.

Conclusions
Using a combined approach of ab initio and Deep Potential Molecular Dynamics (DPMD), Prof. Jun Cheng's team conducted an in-depth investigation into the proton transfer (PT) mechanisms at the rutile SnO₂(110)/H₂O interface. The study identified three distinct types of PT pathways and revealed, through free energy calculations, that the mediated-PT pathway forms the shortest hydrogen bond with the assistance of solvent water molecules, exhibiting the lowest energy barrier and the fastest reaction rate.
Moreover, the study found that a fully solvated environment plays a crucial role in the proton conduction mechanism of the mediated-PT pathway, whereas its impact on direct PT reactions is relatively minor. Finally, the authors summarized different types of PT pathways at oxide/water interfaces, providing molecular-level insights into proton transfer mechanisms relevant to key chemical processes in electrochemistry, photoelectrocatalysis, colloid science, and geochemistry.
This research advances the understanding of PT reaction mechanisms at oxide interfaces and contributes to the development of related scientific fields.