What Can Uni-Mol Do too? | Unveiling NAG2G: A Powerful Tool for Enhancing Single-Step Retrosynthesis Prediction
On February 13, 2024, DP Technology published a cover article in JACS Au titled "Node-Aligned Graph-to-Graph: Elevating Template-free Deep Learning Approaches in Single-Step Retrosynthesis." This study developed a Transformer-based Node-Aligned Graph-to-Graph (NAG2G) model, significantly improving the accuracy of single-step retrosynthesis prediction.
The NAG2G model integrates 2D molecular graph and 3D conformation information, achieving atom mapping between products and reactants through node alignment. This approach overcomes the limitations of traditional template-based methods.
This groundbreaking achievement provides a powerful tool for chemical synthesis design, advancing the field of retrosynthesis and setting a new standard for single-step prediction methodologies.
Research Background
Single-step retrosynthesis (SSR) is a critical step in organic chemistry that involves reverse reasoning to synthesize a target product or intermediate. SSR plays a pivotal role in automated multi-step synthesis route design. With the advancement of computer-aided synthesis planning tools, researchers have turned to deep learning (DL) techniques to address this task.
DL frameworks for SSR can be broadly categorized into three types: template-based, semi-template-based, and template-free.
- Template-based methods rely on existing reaction rules for retrosynthesis design.
- Semi-template-based methods involve a two-step process: identifying the reaction center and then predicting reactants.
- Template-free methods, on the other hand, directly predict reactants using deep learning models.
However, template-free methods often overlook critical 2D molecular information and struggle with atom alignment during node generation, leading to performance limitations compared to template-based and semi-template-based approaches.
To overcome these challenges, this work introduces the Node-Aligned Graph-to-Graph (NAG2G) model, a Transformer-based template-free DL model. NAG2G integrates 2D molecular graph and 3D conformation information, preserving comprehensive molecular details. It employs node alignment to achieve atom mapping between products and reactants, generating the node graph step-by-step in an autoregressive manner.
Through extensive benchmarking and case studies, this work demonstrates the exceptional performance of NAG2G in terms of prediction accuracy and practical applicability.
Methods
Methodology
2.1 Computer-Aided Single-Step Retrosynthesis (SSR) Workflow
Figure 1 outlines the three primary design methods for computer-aided SSR: template-based, semi-template-based, and template-free approaches. Each has its own characteristics and limitations:
Template-Based Methods:
These methods rely on a dictionary of known reaction rules, predicting retrosynthesis pathways by matching reaction templates. While intuitive, their coverage is limited to the template library, making it difficult to predict novel reactions outside the template scope.Semi-Template-Based Methods:
These divide the SSR task into two stages: first detecting synthons or intermediates, and then predicting the reactants. While this expands the search capacity to some extent, errors in each stage can accumulate, negatively impacting the final prediction results.Template-Free Methods:
Models like NAG2G use deep learning to directly predict reactants without relying on predefined templates. NAG2G integrates 2D molecular graphs and 3D conformations, leveraging node alignment to map atoms between products and reactants, significantly improving prediction accuracy.
The workflow highlights NAG2G's innovative and advantageous ability to retain molecular detail and enhance prediction accuracy, overcoming the limitations of traditional template-based methods. This approach provides a flexible and efficient solution, revolutionizing chemical synthesis design tasks.

2.2 The Role of Uni-Mol in NAG2G
Uni-Mol plays a critical role in processing and representing molecular information within the NAG2G model. By combining 2D molecular graphs and 3D conformations, NAG2G retains comprehensive molecular details, with Uni-Mol serving as a key enabler. Specifically, Uni-Mol's encoder transforms the 2D and 3D molecular information into compact numerical representations, allowing the model to better understand and process molecular structures.
The NAG2G encoder, inspired by Uni-Mol, adopts a Transformer architecture to integrate 2D and 3D information. At each timestep, the encoder processes atom features, edge features, and 3D conformation coordinates to generate compact molecular representations. These representations, through node alignment, enable the model to accurately map atom relationships between products and reactants during the graph generation process.

2.3 Data Augmentation and Node Alignment
Figure 3 illustrates the processes of data augmentation and product-reactant alignment:
Data Augmentation:
Product SMILES sequences are randomly generated using RDKit, and input graph node orders are determined accordingly. This randomization increases input diversity, improving the model's robustness.Node Alignment:
Once the product graph node order is determined, reactant graph node orders are generated following a clear and unique rule:- The order of shared atoms in the reactants is aligned with the product atom order.
- The order of non-shared atoms is extracted based on the aligned reactant SMILES sequence, ensuring a unique and consistent node order for reactants.

2.4 Example Workflow of NAG2G Model
Figure 4 demonstrates an example of the NAG2G model's generation process, highlighting how it predicts reactant molecular graph structures from products using node alignment and stepwise graph generation.
Initial Input:
The model takes the product's molecular graph as input, including atom, edge, and 3D conformation information.Node Generation:
At each timestep, the decoder generates a new node based on the current molecular graph structure and previously generated nodes. The node alignment strategy ensures the generated node sequence aligns with the product's node order.Edge Generation:
Along with node generation, the decoder predicts edges connecting new nodes to existing nodes, determining bond types and relationships.Stepwise Reactant Graph Construction:
By repeating the above steps, the model iteratively constructs the complete reactant molecular graph while ensuring accurate alignment with the product graph.

2.5 Decoder Attention Mechanism: Enhancing NAG2G Generation Efficiency
The decoder generates reactant molecular graphs autoregressively. Figure 5 explains how the attention mechanism efficiently handles time-varying graph features at each timestep:
Autoregressive Generation:
At each timestep, the decoder generates a new node based on previously generated nodes, edges, and current graph features. To ensure accuracy, the decoder uses the true output from the previous step as input for the current step.Multi-Head Attention:
Multi-Head Attention is employed to process the query, key, and value matrices in parallel at each timestep. The computational complexity of each attention head is ( n \times n \times d_h ), with peak memory consumption of ( n \times n \times n ).Time-Varying Graph Features:
A time-varying graph feature matrix ( D ) is introduced into the attention mechanism as additive positional encoding. This ensures that graph features at each timestep are correctly utilized during the generation process.Memory Optimization:
To reduce computational costs and memory consumption, Figure 5 illustrates an optimization method that reduces the dimension of matrix ( D ), lowering peak memory usage to ( n \times n \times d_h^2 ). This makes the attention mechanism more efficient for real-world applications.
The decoder's attention mechanism ensures efficient and accurate graph generation, leveraging dynamic graph features at each timestep. This design enables the model to capture temporal dependencies during reactant graph construction, achieving high-precision single-step retrosynthesis prediction.

3.Results and Discussion
3.1 Model Evaluation
Performance on USPTO-50k Dataset

Table 1 presents the Top-k accuracy of the NAG2G model on the USPTO-50k dataset, compared against other existing models, including template-based, semi-template-based, and template-free approaches. Results show that NAG2G outperforms all other models across all evaluation metrics, particularly excelling in Top-1 and Top-10 accuracy.
Performance on USPTO-Full Dataset

Table 2 highlights the Top-k accuracy of NAG2G on the larger-scale USPTO-Full dataset. Despite the increased task complexity compared to USPTO-50k, NAG2G consistently demonstrates strong predictive capabilities, outperforming most existing methods.
Ablation Study
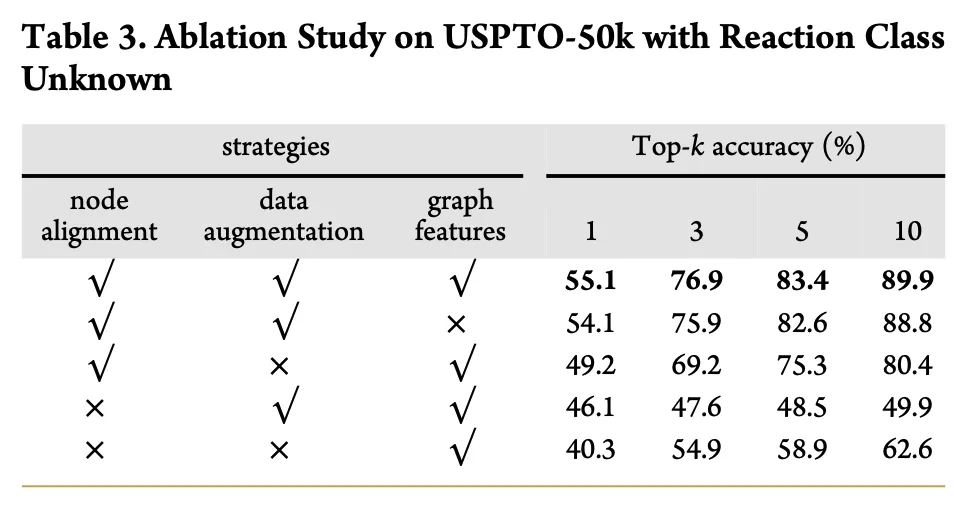
Table 3 presents an ablation analysis that explores the importance of various components in the NAG2G model, such as node alignment, data augmentation, and graph features. By systematically removing these components, the study evaluates their impact on model performance. Results indicate that node alignment and data augmentation are crucial for improving prediction accuracy.
Molecular Validity

Table 4 evaluates the Top-k validity of molecules generated by NAG2G on the USPTO-50k dataset. The results demonstrate NAG2G's exceptional performance in generating valid molecules, further verifying its reliability in molecular generation tasks.
3.2 Practical Application Cases: Exceptional Performance of NAG2G in Drug Molecule Retrosynthesis Prediction
Figure 6 showcases the retrosynthesis pathways for four drug molecules (Nirmatrelvir, Osimertinib, Salmeterol, and Lenalidomide) and provides detailed examples of NAG2G’s performance in predicting these pathways. These cases validate the accuracy and efficacy of NAG2G in real-world applications.
Nirmatrelvir:
- The figure displays a six-step synthesis pathway for Nirmatrelvir.
- NAG2G successfully predicted all reaction steps, with the top-ranked predictions for the first three steps, highlighting its outstanding performance in predicting complex reaction pathways.
Osimertinib:
- Osimertinib, a drug used to treat non-small-cell lung cancer, features a five-step retrosynthesis pathway.
- NAG2G accurately predicted each reaction type, with most predictions ranking first, showcasing its efficiency in drug molecule retrosynthesis.
Salmeterol:
- The synthesis pathway for Salmeterol consists of five steps.
- NAG2G accurately predicted key steps such as protection reactions, etherification, and asymmetric Henry reactions, demonstrating its flexibility and accuracy when handling molecules with complex functional groups.
Lenalidomide:
- Lenalidomide is a complex drug molecule with a four-step synthesis pathway.
- NAG2G successfully predicted reduction reactions and NBS (N-Bromosuccinimide) substitution reactions and proposed a reasonable cyclization mechanism, further showcasing its capability to predict complex reactions.
Figure 6 underscores NAG2G's advantages in high-precision prediction and multi-step reaction handling by demonstrating its application in the synthesis pathways of real-world drug molecules. These examples validate the model's reliability and accuracy while highlighting its tremendous potential in drug discovery and chemical synthesis design.

Conclusions
This study introduces the NAG2G model, a template-free deep learning approach based on node alignment and a Transformer architecture for single-step retrosynthesis prediction. By integrating 2D molecular graphs and 3D conformation information, NAG2G preserves detailed molecular structure features and achieves precise product-to-reactant atom mapping through its node alignment strategy.
The Uni-Mol encoder plays a critical role in the NAG2G model. By converting 2D and 3D information into efficient numerical representations, Uni-Mol enables the model to better understand and process molecular structures, significantly improving prediction accuracy.
Experimental results demonstrate that NAG2G outperforms existing template-based and semi-template methods on both the USPTO-50k and USPTO-Full datasets, showcasing exceptional predictive performance.
Ablation studies further validate the importance of node alignment, data augmentation, and time-varying graph features in enhancing model performance. By leveraging the Uni-Mol encoder, NAG2G efficiently handles and accurately predicts complex chemical reactions.
Real-world case studies highlight NAG2G’s powerful capabilities in predicting synthesis pathways for drug molecules, confirming its vast potential and practical applicability in chemical synthesis design.